Главная страница Случайная страница
КАТЕГОРИИ:
АвтомобилиАстрономияБиологияГеографияДом и садДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеталлургияМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРелигияРиторикаСоциологияСпортСтроительствоТехнологияТуризмФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Молекула монооксида углерода
|
|
Гетероатомная молекула оксида углерода имеет очень большую энергию связи 256 кКал/моль. Она слабый донор и сильный p-акцептор на разрыхляющие орбитали. Относительное расположение молекулярных орбиталей для СО имеет особенность. Pz уровень в атоме кислорода расположен ниже, чем соответствующий уровень для атома углерода (из-за большего заряда ядра), поэтому он (Pz уровень в атоме О) сильно взаимодействует с 2S–орбиталью атома углерода. В связи с этим Pz–молекулярная орбиталь СО располагается выше, чем вырожденные Pх и Pу орбитали (см. диаграмму). Пара электронов, которая располагается на Pz орбитали, является несвязывающей и локализована на атоме углерода, а пара электронов на 2S–орбитали - также несвязывающая и локализована на атоме кислорода. Причем, Pz – молекулярная орбиталь имеет в основном р-характер с большим лепестком, вытянутым от связи С-О. Вакантные орбитали (Pz*, Pх*, Pу*) также локализованы на атоме углерода, поэтому практически во всех случаях оксид углерода координируется через углерод.
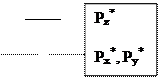 | ||||||
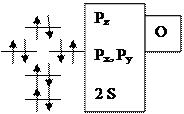 | ||||||
| ||||||
Рис. 5. Диаграмма относительного расположения МО в молекуле оксида углерода(II)
За счет заполненной Pz-орбитали, локализованной на углероде, молекула СО обладает слабыми донорными свойствами и образует донорно-акцепторную компоненту связи, взаимодействуя с подходящей по симметрии вакантной орбиталью dz2 переходного металла. За счет вакантных разрыхляющих Px и Py орбиталей у СО есть возможность проявлять акцепторные свойства. Две разрыхляющие p-орбитали по симметрии могут взаимодействовать с заселенными dxy и dxz орбиталями переходного металла.
Из всего вышесказанного следует, что СО является s-лигандом, но в подавляющем большинстве случаев его следует рассматривать одновременно как s-донор и p-акцептор с преобладанием акцепторных свойств.
Карбонильные комплексы известны для большинства переходных металлов. Первые комплексы были получены в конце 19-го века. Например [Pd(CO)X]n, [Pt(CO)X]n, Co2(CO)8, Ni(CO)4. Координация оксида углерода в комплексах бывает концевая (терминальная) и мостиковая (с участием двух атомов металла или трех атомов металла).

Первый тип координации является концевым и реализован во многих моно- и полиядерных комплексах, например в тетраэдрическом моноядерном тетракарбониле никеля или биядерном дикобальтоктакарбониле.
Следующие типы координации - симметричная и несимметричная мостиковая (µ- и µ3-тип) - широко распространены в химии кластеров. Так, в кластере состава Pd4(CO)4(OAc)4 (рис. 3) обнаружены только µ-СО-группы, а в кластере [Pt3(CO)6]2-n – и концевые, и мостиковые лиганды СО.
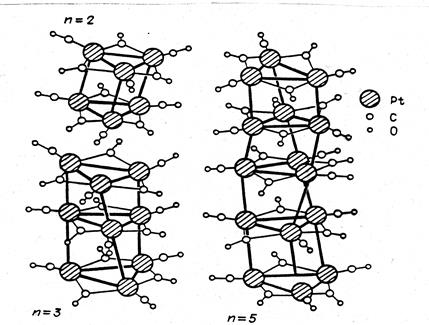
Рис. 6. Карбонильные кластеры платины [Pt3(CO)3(µ-CO)3]2-n
Следует добавить, что при терминальной координации карбонил является донором 2 электронов. Эта же донорная способность сохраняется в том случае, если СО координирован мостиком, но при этом направление связи С-О остается перпендикулярным ребру (или грани), на которой он координирован. В противном случае в связь с металлом начинают включаться электроны кратной связи и атома кислорода.
Как уже отмечалось выше, координация СО по обоим атомам возможна, хотя и встречается относительно редко. При такой координации происходит значительное удлинение связи С-О (от 1.13 до 1.30 А). Примеры такой координации приведены ниже.
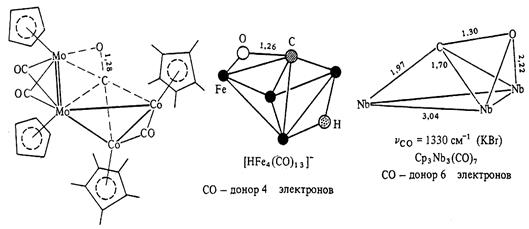
Если в образовании связи металл-лиганд участвуют оба атома, то СО-группа является донором 4 электронов. В случае, когда молекула СО расположена параллельно плоскости М3-цикла, она становится донором 6 электронов.
И, наконец, линейная координация СО отмечена в металлоорганических соединениях металлов начала больших периодов периодической системы:
(CO)5V-C-O-V-O-C-V(CO)5 или (η 5-С5Me5)(Me)Ti-O-C-Mo(CO)3Cp
 Координационная связь между СО и переходным металлом складывается из двух компонент – донорноакцепторной (СО – донор, металл – акцептор за счет вакантных орбиталей, например, dz2 и dx2-y2 для октаэдрических комплексов) и дативной (металл – донор за счет заполненных dxy, dxz, dyz, СО – акцептор за счет вакантных орбиталей. Обе компоненты способствуют ослаблению связи С – О. В зависимости от степени заселения разрыхляющих орбиталей СО происходит большее или меньшее увеличение длины, понижение частоты валентных колебаний и уменьшение энергии связи С – О (табл. 5).
Координационная связь между СО и переходным металлом складывается из двух компонент – донорноакцепторной (СО – донор, металл – акцептор за счет вакантных орбиталей, например, dz2 и dx2-y2 для октаэдрических комплексов) и дативной (металл – донор за счет заполненных dxy, dxz, dyz, СО – акцептор за счет вакантных орбиталей. Обе компоненты способствуют ослаблению связи С – О. В зависимости от степени заселения разрыхляющих орбиталей СО происходит большее или меньшее увеличение длины, понижение частоты валентных колебаний и уменьшение энергии связи С – О (табл. 5).
Из приведенных данных видно, что при координации происходит изменение свойств молекулы СО (см. выше) и координированная молекула по характеристикам приближается к карбонильной группе органических соединений. А в этом и заключается цель большинства химических процессов органического синтеза с участием СО. Изменение характеристик координированного СО приводит к изменению реакционной способности.
Таблица 5
Характеристики молекулы монооксида углерода и группы СО в различных состояниях
| Молекула или ион | Энергия связи С-О, кКал/моль | Частота валентных колебаний, см-1 | Длина связи С-О, Å |
| СО (некоорд. молекула) | 1, 128 | ||
| СО+ | 1, 115 | ||
| СО (коорд. концев.) | 2214 ¸ 1980 | ||
| СО (мостиков.) | 1970 ¸ 1650 | ||
| Ni(CO)4 | 1, 15 | ||
| H2CO | 1, 21 |
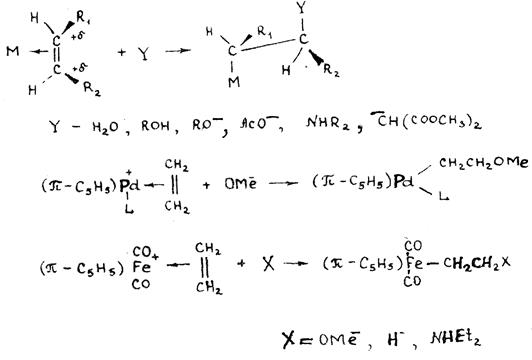
Наиболее характерная реакция координированного СО – реакция с нуклеофилами с образованием ацильного металлоорганического соединения. Образование интермедиатов
-М-Сº О + У- ® -М-- С-У
║
О
такого типа предполагается в процессах карбонилирования алкинов, окислительного карбонилирования спиртов, алкенов и алкинов.
Изложенная схема активации применима к большинству субстратов: алкенам, диенам, алкинам, ароматическим соединениям, СО2.
2.2.3 Активация алкенов и алкинов
Превращения алкенов и алкинов относятся к важнейшим реакциям основного органического синтеза. Как правило, эти превращения включают активацию π -лигандов в π -комплексах переходных металлов.
π -Комплексы моноолефинов, сопряженных и несопряженных диолефинов и алкинов известны почти для всех переходных металлов. При этом встречаются моно-, би- и полиядерные соединения. Известны также соединения, содержащие 2, 3 и даже 4 молекулы алкена на атом металла, например, AcacRh(C2H4)2, Ni(C2H4)3 и Ir(C2H4)4Cl. В π -комплексах алкинов состава Mm(C2R2)n соотношение m/n меняется весьма широко. Например, известны комплексы состава Co4(CO)10(C2H2), Pt(C2R2)2, W(CO) (C2R2)3.
Координация всех π -лигандов сопровождается более или менее значительными изменениями их физических характеристик – понижается частота валентных колебаний кратных связей, увеличивается длина связей С-С, изменяются величины валентных углов. Природа химической связи в π -комплексах переходных металлов имеет много общего.
Рассмотрим основные положения на примере π -комплексов алкенов и алкинов. Молекула этилена по величине потенциала ионизации не отличается от молекулы аммиака (IC2H4 = 10.5 эв). Донорные свойства ацетилена выражены несколько слабее (IC2H2 = 11.4 эв). В ацетилене, однако, нижние вакантные МО лежат ниже, чем у этилена, поэтому молекула ацетилена характеризуется более выраженными акцепторными свойствами.
Представления о природе связи в π -комплексах базируются на идеях Дьюара, Чатта и Данкансона, развитых более полувека назад. Модель Дьюара-Чатта-Данкансона, несмотря на ее упрощенность, может быть положена в основу концепции активации π -лигпндов в комплексах переходных металлов.
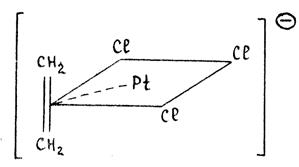
Рассмотрим, например, соль Цейзе KPtCl3(C2H4). Молекула этилена занимает координационное место в плоско-квадратном комплексе PtCl42-, вытесняя ион хлора:
Гибридная орбиталь Pt(II) (например, dsp2-орбиталь) перекрывается с π -МО этилена, образуя трехцентровую двухэлектронную связь (донорно-акцепторная компонента связи). Гибридные заполненные 6dp-орбитали платины взаимодействуют с разрыхляющей π *-МО олефина, образуя вторую трехцентровую двухэлектронную связь (дативная компонента связи). В общем виде эти ситуация представлена на диаграмме:
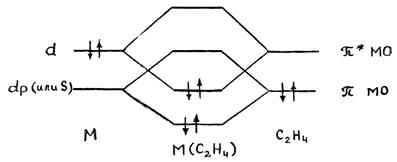
Обе трехцентровые связи вносят вклад в повышение прочности связи M-C2H4. Однако эти компоненты по разному влияют на свойства координированного π -лиганда.
В частности, образование дативной связи увеличивает заселенность разрыхляющей орбитали π -лиганда, что приводит к сильной дестабилизации координированной молекулы. Соотношение двух компонент связи металл-лиганд будет зависеть от свойств центрального атома, других лигандов и заместителей в молекуле π -лиганда. По степени изменения свойств π -лиганда все π -комплексы делятся на две группы.
I тип – π -комплексы, в которых изменение π -лиганда носит характер слабого возмущения.
II тип – π -комплексы, в которых сильно меняются порядок связи С-С и валентные углы.
Комплексы I типа обычно образуются металлами в высоких степенях окисления, связанных с электроотрицательными лигандами. Комплексы II типа образуют металлы в низких степенях окисления, связанные с мягкими основаниями. Так, например, соль Цейзе – K[PtCl3(C2H4)] – является примером комплекса I типа, а соединение (Ph3P)2Pt(C2H4) – примером комплекса II типа (табл. 6).
Таблица 6
Характеристики π -комплексов этилена первого и второго типа
| Комплекс | Тип | Δ ν С=С, см-1 | LC=C, Ǻ | Положение С=С |
| K[PtCl3(C2H4)] (Ph3P)2Pt(C2H4) | I II | > 200 | 1.37 1.43 | Перпендикулярно пл. PtCl3 В плоскости Pt(PPh3)2 |
В комплексах c сильными электроотрицательными π -лигандами длины связей С-С приближаются к длине простой связи С-С в алканах. Например, в комплексе L2Pt(C2F4) длина связи С-С составляет 1.54 Ǻ, т.е. двойная связь по существу превращается в 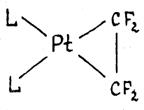 одинарную. Значения химсдвигов 19F и констант спин-спинового взаимодействия в спектре ЯМР комплекса также хорошо объясняются “циклопропановой” структурой. Таким образом, взаимодействие сильного донора L2Pt (0) с сильным акцептором C2F4 приводит к существенному переносу d-электронов на π -лиганд и образованию металлоцикла. Происходит окислительное присоединение π -лиганда к Pt(0) с образованием Pt(II) и аниона C2F42-. Особенно отчетливо различие двух групп π -комплексов можно проследить в случае алкиновых лигандов (табл. 7).
одинарную. Значения химсдвигов 19F и констант спин-спинового взаимодействия в спектре ЯМР комплекса также хорошо объясняются “циклопропановой” структурой. Таким образом, взаимодействие сильного донора L2Pt (0) с сильным акцептором C2F4 приводит к существенному переносу d-электронов на π -лиганд и образованию металлоцикла. Происходит окислительное присоединение π -лиганда к Pt(0) с образованием Pt(II) и аниона C2F42-. Особенно отчетливо различие двух групп π -комплексов можно проследить в случае алкиновых лигандов (табл. 7).
Таблица 7
Характеристики π -комплексов алкинов
| Комплекс | Тип | Δ ν С=С, см-1 | LC=C, Ǻ | Положение С=С |
| (Bu2C2PtCl2L* (Ph2C2)Pt(PPh3)2 | I II | 1.27 1.36 | Перпендикулярно пл. PtCl3 В плоскости Pt(PPh3)2 |
* L - амин
2.2.3.1 Реакции координированных алкенов
Наиболее характерной реакцией π -комплексов олефинов, относящихся к I типу, является реакция с нуклеофильными реагентами. Нуклеофильный реагент часто атакует π -комплекс со стороны, противоположной металлу (транс-присоединение), образуя σ -металлоорганическое соединение:
В качестве нуклеофилов могут выступать анионные карбонилметаллаты, образуя σ -металлоорганические соединения с мостиковым этиленом:
(π -C5H5)(CO)3M(π -C2H4) + Re(CO)5- → (π -C5H5)(CO)3MCH2CH2Re(CO)5
M = Mo, W
Поляризация молекулы олефина в π -комплексе может приводить к смещению электронов в группах, соседних с С=С – связью, что проявляется, например, в образовании π -аллильных комплексов:
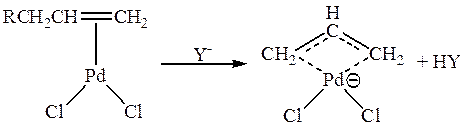
В π -комплексах II типа характер реакционной способности кратной связи меняется. π -Лиганд становится способным взаимодействовать с электрофильными реагентами, например в соответствии со схемой:
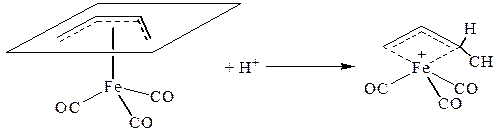
Более характерными для π -комплексов II типа являются реакции, связанные с общим разрыхлением всей молекулы из-за переноса электронов на олефин. К таким реакциям следует отнести реакции циклообразования, внедрения по связи металл-металл, окислительного присоединения или замещения по связи =С-Х (где X = H, Cl, F).
Например:
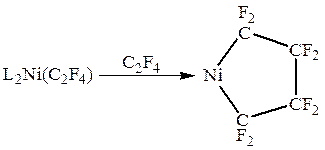
Или
(CO)4Co-Co(CO)4 + CF2=CF2 → (CO)4Co- CF2CF2-Co(CO)4
Иногда активация олефина в π -комплексах II типа настолько лабилизирует связи в π -лиганде, что становится возможным разрыв связей С-Н при двойной связи и в соседней с двойной связью метильной группе, приводящий к продуктам окислительного присоединения:
Os3(CO)12 + C2H4 → H2Os3(C=CH2)(CO)9 + 3CO
2.2.3.2 Реакции координированных алкинов
В комплексах алкинов типа I (включающих Ag(I), Cu(I), Hg(II), Pt(II), Pd(II), Ru(III) и др.) повышение электрофильности тройной связи приводит к облегчению взаимодействия с нуклеофильной частицей из раствора (транс -присоединение) или нуклеофилом, координированным металлом (цис -внедрение):
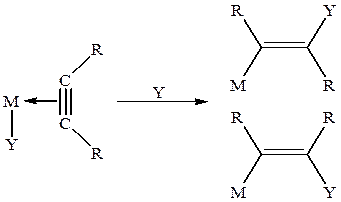
Основные реакции π -комплексов I типа довольно удачно промоделированы на комплексах Pt(II):

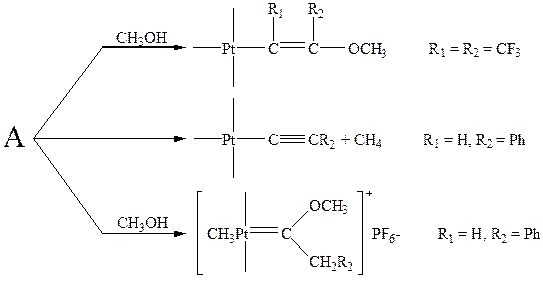
Образующийся катионный π -комплекс (А) способен превращаться по нескольким направлениям. Первая реакция типичное для π -комплексов первого типа присоединение нуклеофила с образованием β -замещенного винильного производного металла. Вторая реакция протекает в результате электрофильного замещения протона ионом CH3PtL2+ c последующим электрофильным замещением L2PtC≡ CR2 ионом Н+ у атома углерода метильной группы.
В ходе третьей реакции происходит перегруппировка алкин → винилиден с образованием карбенового комплекса CH3PtL2(=C=CHR2) c последующим присоединением нуклеофила (СН3ОН) к карбеновому атому углерода и образованием метоксикарбенового лиганда.
В π -комплексах II-го типа в первую очередь сильно разрыхляется тройная связь углерод-углерод и связь C-X при тройной связи. Так, активация связи С-Н в π -комплексах Ni(0), Pt(0), Os(0) или Rh(I) приводит к окислительному присоединению с образованием этинилгидридного комплекса металла:
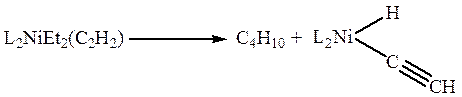

Вторая характерная реакция для π -комплексов II типа – это реакция циклообразования, причем в состав получающегося металлоцикла входят уже две молекулы алкина:
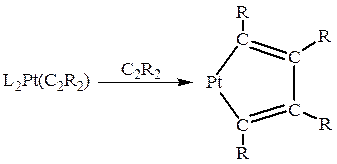
Металлоциклопентадиены из алкинов получены в реакциях комплексов Pt(0), Pd(0), Co(I), Fe(0), Rh(I), Ir(I) и Ti(II).
2.2.4 Активация полярных молекул
Полярные молекулы НХ, где Х – ОН, OR, Hal, CN, NH2, NR2, NHR, SR и др. активируются по механизму, близкому к механизму активации апротонными кислотами. Образование донорно-акцепторной связи между донорным атомом полярной молекулы и комплексом переходного металла, имеющим вакантные орбитали, приводит к ослаблению связи Н – Х. Ослабление связи H-X при координации этих молекул подтверждается, как правило, данными ИК-спектров координированных молекул. При этом в образующемся комплексе происходит смещение σ -пары электронов донорных атомов O, N или S к иону металла, обладающему акцепторными свойствами. Молекула лиганда поляризуется, что приводит к ее ионизации и облегчает диссоциацию (в полярных растворителях):
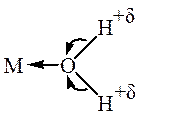
Смещение электронов и ослабление связи Х-Н при координации подтверждается данными ИК-спектров координированных молекул. Например, координация RNH2 в комплексах с PtCl2 приводит к понижению ν N-H (на 80-100 см-1). В результате повышается способность связи Н-Х к гетеролитической диссоциации с передачей протона на растворитель, на другой субстрат или его окислительному присоединению к переходному металлу (в зависимости от степени окисления металла и состояния его внутренней координационной сферы).
HX + MLn —→ H--X→ MLn-1 + L
Даже такие слабые кислоты, как молекула аммиака или аминов, легко депротонируются в водных или неводных средах в координационной сфере переходных металлов:
TiCl4 + 6NH3 → TiCl(NH2)3 + 3 NH4Cl
Или
Pt(NH3)X5- + H2O → Pt(NH2)X52- + H3O+
В результате в комплексе металла появляется фрагмент молекулы HX (например, NH2), реакционная способность которого, конечно, ниже, чем свободного иона NH2-, но концентрация которого на много порядков выше, чем в отсутствие комплексообразователя. Константа диссоциации циановодорода HCN, например, составляет 10-10. Образование комплексов с металлами позволяет существенно повысить концентрацию группы M-CN. Так, при взаимодействии HCN с полиядерными комплексами меди(I) концентрация CuCN в
CumCln(n-m)- + HCN → ClnCum-1(CuCN) + HCl
растворе может достигать 15% масс. Координированный анион CN может далее участвовать в различных реакциях с образованием изонитрила CH3N=C, β -цианвинильного производного Cu(I) или изомера цианистого водорода NH=C, быстро присоединяющего нуклеофил Х- с образованием имина в координационной сфере металла:
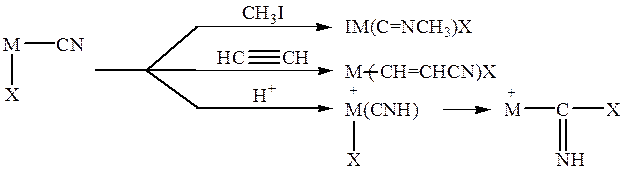
Для катализа особенно важны две последние реакции.