Главная страница Случайная страница
КАТЕГОРИИ:
АвтомобилиАстрономияБиологияГеографияДом и садДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеталлургияМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРелигияРиторикаСоциологияСпортСтроительствоТехнологияТуризмФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Успадкування аутосомно-рецесивних ознак у людини
|
|
На відміну від аутосомно-домінантного типу успадкування аутосомно-рецесивне виявляється лише в результаті шлюбу двох гетерозигот. Тому такі ознаки частіше з’являються у близькородинних шлюбах. Чим менша концентрація аутосомно-рецесивного гена в популяції, тим більша вірогідність його реалізації при кровній спорідненості батьків. За аутосомно-рецесивним типом успадковуються більше 780 хвороб.
Деякі аутосомно-рецесивні ознаки наведено в табл. 1.
За аутосомно-рецесивним типом успадковується ген, що зумовлює здатність сприймати смак фенілтіокарбаміда або схожих з ним сполук.
Рецесивним є ген, що приводить до порушення будови внутрішнього вуха і, отже, до глухоти. Відсутність слуху з самого народження веде до німоти. Ген цей зустрічається досить часто, і шлюби між двома глухонімими — поширене явище. Цікаво відзначити, що від такого шлюбу іноді народжуються, всупереч сподіванням, здорові (з нормальним слухом) діти.
За аутосомно-рецесивним типом успадковуються багато захворювань з порушеннями обміну речовин. Ця велика група захворювань налічує близько 600 ензимопатій: відсутність або недостатність відповідного ферменту приводить до блокування біохімічних реакцій. Ензимопатії виникають в результаті мутацій генів, відповідальних за синтез певного ферменту.


 У більшості випадків ензимопатії протікають паралельно з розумовою відсталістю. Виключенням із загального правила є одне з порушень амінокислотного обміну — альбінізм — відсутність ферменту тиразинази, яка перетворює амінокислоту тирозин в пігмент меланін. У окремих індивідуумів меланін зовсім не утворюється або утворюється в таких малих кількостях, що шкіра, волосся виявляються дуже світлими, а очі — червоними (відсутність пігменту не маскує кровоносні судини сітківки). Світлобоязнь і незвичайний колір очей примушують таких людей носити темні окуляри. В Екваторіальній Америці збереглося ціле плем’я альбіносів.
У більшості випадків ензимопатії протікають паралельно з розумовою відсталістю. Виключенням із загального правила є одне з порушень амінокислотного обміну — альбінізм — відсутність ферменту тиразинази, яка перетворює амінокислоту тирозин в пігмент меланін. У окремих індивідуумів меланін зовсім не утворюється або утворюється в таких малих кількостях, що шкіра, волосся виявляються дуже світлими, а очі — червоними (відсутність пігменту не маскує кровоносні судини сітківки). Світлобоязнь і незвичайний колір очей примушують таких людей носити темні окуляри. В Екваторіальній Америці збереглося ціле плем’я альбіносів.
До порушень амінокислотного обміну відносяться також алкаптонурія, фенілкетонурія й інші «природжені дефекти метаболізму».
При алкаптонурії у хворих виділяється темна, майже чорна сеча. Пов’язано це з присутністю в сечі гомогентензинової кислоти — проміжного продукту метаболізму двох амінокислот — фенілаланіна та тирозина. У хворих забарвлені також хрящі і в літньому віці розвиваються артрити (від грец. артрон — суглоб).
Фенілкетонурія (хвороба Феллінга) — різке підвищення змісту в крові амінокислоти фенілаланіна і перетворення її в ряд продуктів, наприклад у фенілпіровиноградну і фенілмолочную кислоти. На відміну від гомогентензинової кислоти, яка не має явного несприятливого впливу на тканини мозку, продукти, що утворюються при фенілкетонурії, є дуже токсичними. Тому у дітей при цій патології спостерігається різко виражена розумова відсталість. Захворювання виражається також у зниженні кількості пігменту меланіну, тому хворі завжди виглядають, як блакитноокі блондини зі світлою шкірою. В даний час діагноз можна поставити при народженнідитини експрес-методом: на змочену сечею пелюшку наносять 5 крапель 10% розчину FеС1з, при цьому спостерігається почервоніння, яке швидко проходить).
З наймолодшого віку з їжі таких дітей слід виключити фенілаланін, рекомендується спеціальна дієта: каші на кобилячому молоці (бідному на амінокислоти), мед, масло, овочі, саго (фенілаланіну зовсім не містять) у поєднанні з лікуванням берлофеном (гідролізат білка). Цим попереджають розвиток порушень функцій мозку і діти розвиваються нормально. Без лікування у новонароджених при звичному годуванні розвивається ряд розладів, що приводять надалі до передчасної смерті. В деяких популяціях частота цього гена складає 1: 7000. В 15% випадків це захворювання зареєстровано у нащадків, що походять від шлюбу кровно споріднених батьків. У плазмі крові таких гетерозигот спостерігається вдвічі більше норми фенілаланіна після стандартної дієти із звичним вмістом у ній цієї амінокислоти.
 Порушення ліпідного обміну — амавротична ідіотія (хвороба Тея-Сакса), пов’язана з відсутністю ферменту гексосаміндази А, що спричиняє важкий розлад нервової системи. Цю хворобу можна виявити лише в другій половині першого року життя дитини, коли спостерігається прогресуюче відставання фізичного розвитку, порушення зору й інтелекту, надалі хворий сліпне, розвивається недоумство і повна безпорадність. Важкі симптоми наростають, приводять до смерті дитини у віці до 4–5 років.
Порушення ліпідного обміну — амавротична ідіотія (хвороба Тея-Сакса), пов’язана з відсутністю ферменту гексосаміндази А, що спричиняє важкий розлад нервової системи. Цю хворобу можна виявити лише в другій половині першого року життя дитини, коли спостерігається прогресуюче відставання фізичного розвитку, порушення зору й інтелекту, надалі хворий сліпне, розвивається недоумство і повна безпорадність. Важкі симптоми наростають, приводять до смерті дитини у віці до 4–5 років.
Це захворювання рідкісне, зустрічається з частотою 1: 250000 часто виявляється у подружжя, предками якого були вихідці з Бухари або інших областей Середньої Азії.
Галактоземія — порушення вуглеводневого обміну. Вона обумовлена порушенням діяльності печінки, накопиченням у тканинах (у тому числі і в крові) галактози, яка є у молоці. Без лікування розвивається цироз печінки; до патологічного процесу залучаються й інші життєво важливі органи. Зрештою хвороба приводить до недоумства і ранньої смерті. На початку життя, як тільки новонароджений починає одержувати молоко, спостерігаються жовтяниця, блювота, диспепсичні розлади, зниження маси тіла. При ранній діагностиці таких дітей до трирічного віку переводять на безмолочне вигодовування, тобто виключають продукти, що містять галактозу. Такі діти розвиваються нормально і відхилень у психіці у них не спостерігається. Носіями такого гена, що викликає захворювання (тобто гетерозиготи) є в середньому 1: 70000.
Аномалії, пов’язані з порушеннями розпаду деяких вуглеводовмісних сполук, викликають розвиток мукополісахаридозів (гаргоїлізми). При цих захворюваннях уражена сполучна тканина, а отже, страждають опорно-трофічні функції і моторика. Для хворих мукополісахаридозом характерна потворна статура (діти нагадують потвор-гаргоїдів, які прикрашають паризький храм Нотр-Дам), наявність множинних вад внутрішніх органів (печінки, нирок, серця, аорти, нервової системи) і очей.
 Надзвичайно рідкісне спадкове злоякісне захворювання — природжений іхтіоз (від грец. іхтіс — риба). Вся шкіра такого хворого покрита значними за розміром зроговіли пластинами, що нагадують луску риби; при цьому порушенні є неможливим шкірне дихання. Дитина або народжується мертвою, або вмирає незабаром після народження.
Надзвичайно рідкісне спадкове злоякісне захворювання — природжений іхтіоз (від грец. іхтіс — риба). Вся шкіра такого хворого покрита значними за розміром зроговіли пластинами, що нагадують луску риби; при цьому порушенні є неможливим шкірне дихання. Дитина або народжується мертвою, або вмирає незабаром після народження.
Як приклад злоякісних новоутворень, що передаються по аутосомно-рецесивному типу, можна назвати також гліому сітківки ока — пухлину глії (проміжної тканини центральної нервової системи).
Успадкування ознак людини, зчеплених зі статтю
Стать людини залежить від наявності певного поєднання статевих хромосом: XX — у жінок, XY — у чоловіків.
Розвиток первинних (статеві залози і зовнішні статеві органи) і вторинних статевих ознак чоловічої статі визначають гени, розташовані в Y-хромосомі. При відсутності Y- і наявності Х-хромосоми розвивається особина, що названа інтерсексом (від лат. intег — між, sexus — стать) — істота з проміжною статтю (синдром Шерешевського–Тернера), а людина, що має дві або більше Х-хромосоми, за наявності Y-хромосоми буде особиною чоловічої статі (синдром Кляйнфельтера).
Х- і Y-хромосоми мають загальні гомологічні ділянки. В цих ділянках локалізовані гени, що детермінують ознаки, що успадковуються однаково як у чоловіків, так і у жінок. Як приклад захворювання подібного типу можна назвати рецесивний злоякісний новоутвір — пігментну ксеродерму (надчутливість до ультрафіолетового проміння, під впливом якого на відкритих частинах тіла з’являються плями, що пігментуються, — спочатку у вигляді ластовиння, а потім все більших папілом (родимок на ніжках) різної величини і форми і, нарешті, пухлин. Для 2/3 хворих пігментна ксеродерма закінчується смертю до настання статевої зрілості. Іншим прикладом може служити хвороба Огуті (часто зустрічається в Японії); захворювання виражається в появі пігментного ретиніту і аномалії розвитку сітківки. Рецесивний (а іноді і домінантний) характер успадкування проявляють спастична параплегія (порушення діяльності і слабкість нижніх кінцівок, що виникає в результаті дегенерації пірамідальних шляхів стовбурної частини спинного мозку) і епідермальний булльоз (утворення міхурів після механічних травм шкіри).
Окрім гомологічних ділянок, Х- і Y-хромосоми мають негомологічні ділянки. Негомологічна ділянка Y-хромосоми, окрім генів, що визначають чоловічу стать, містить гени перетинок між пальцями і волохатих вух. Патологічні ознаки, зчеплені з негомологічною ділянкою Y-хромосоми, передаються всім синам, оскільки вони одержують від батька Y-хромосому. Міжпальцеві перетинки було знайдено в чотирьох поколіннях однієї сім’ї і лише у синів.
Негомологічна ділянка Х-хромосоми містить в своєму складі цілий ряд рецесивних (для жінок) і домінантних генів.
Прикладом такого роду успадкування є гемофілія; агаммаглобулінемія, нецукровий діабет, дальтонізм і облисіння.
Під загальною назвою «гемофілія» зібраний ряд спадкових захворювань, пов’язаних із зниженням здатності крові згортатися. Гемофілія може розвинутися в результаті нестачі антигемофільного глобуліну (при цій формі захворювання хворим вводять плазматичний екстракт, що містить антигемофілітичний чинник).
Поранення, навіть подряпина або удар можуть викликати сильні зовнішні або внутрішні кровотечі, які нерідко закінчуються смертю. Тому хворих на гемофілію дітей слід ретельно оберігати від всякого роду травм. В деяких країнах (наприклад, у Франції) для таких дітей створені спеціальні школи.
Деякі форми гемофілії успадковуються по аутосомно-домінантному і аутосомно-рецесивному типам. Найпоширеніша форма цього серйозного захворювання зчеплена зі статтю. Оскільки передача гемофілії пов’язана з Х-хромосомою, ця хвороба зустрічається у чоловіків. У жінок вона практично не спостерігається, хоча описані окремі випадки.
Гемофілія здобула широку популярність у зв’язку з тим, що ця патологія спостерігалася серед членів декількох царюючих сімей Європи. Виникнувши, мабуть, як мутація у англійської королеви Вікторії, гемофілія зустрічалася в Іспанії, Німеччині та Росії.
Діабет нецукровий — гіпофункція гіпофіза — приводить до різкого зневоднення організму, яке у новонароджених гальмує ріст, різко порушує психіку, викликає летальний результат.
Порушення кольорового зору — дальтонізм — знайшов у себе і описав англійський природодослідник Джон Дальтон у 1794 р. Порушення кольорового зору у машиніста, що привело до важких наслідків, було описано в 1875 р. (в Швейцарії, де сталася аварія потягу з великим числом жертв). Цей трагічний випадок послужив приводом для обов’язкової перевірки кольорового зору у працівників всіх видів транспорту, солдатів і ін.
Розрізняють декілька форм дальтонізму: дейтеронопію — часткова аномалія сприймання зеленого кольору (змішують зелений колір з сірим, жовтим і темно-червоним) і протанопію — аномалія сприйняття червоного кольору (змішують червоний колір з сірим, жовтим і темно-зеленим).
Прикладами домінантних, повністю зчеплених зі статтю по Х-хромосомі (тобто зчеплених з негомологічною ділянкою цієї хромосоми) можуть служити дві рідкісні ознаки: гіпофосфатемічний рахіт, який не піддається вітамінотерапії, і ознака відсутності різців у щелепах.
Рецесивні, зчеплені з негомологічними ділянками Х-хромосоми ознаки виявляються значно частіше у чоловіків (внаслідок їх гемізиготності), ніж у жінок. У жінок дана патологія проявиться за у мови наявності двох генів, одержаних від обох батьків.
У минулому столітті, ще до відкриттів Менделя, були вивчені випадки передачі спадкових ознак через цитоплазму.
Відомо, що мітохондрії містять ДНК, що дозволяє говорити про так звані плазмогени, які забезпечують передачу спадкової інформації через цитоплазму яйцеклітини (заплідненні сперматозоїди вносять в зиготу лише ядро). В даний час відомий ряд ознак, які передаються через матір (але не через батька). Прикладом таких захворювань можна назвати спадкову сліпоту, яка виникає в результаті атрофії зорового нерва (синдром Лебера).
Генні і хромосомні мутації у людини
Як приклад хромосомних мутацій приведемо делецію — синдром 5 р — втрата короткого плеча (р) 5-ї хромосоми, або синдром «котячого крику» (назва обумовлена схожістю плачу дитини з нявканням кішки).
Такий крик пояснюється не аномалією голосового апарату, а порушеннями центральної нервової системи. Для синдрому 5 р характерні мікрогнатія (від грец. гнатос — щелепа) і синдактилія, які доповнюють фенотипічну картину синдрому. У хворих спостерігається пониження опору до інфекцій, тому вони вмирають рано. Обтяжуючим чинником є різні порушення внутрішніх органів (аномалії серця, нирок, грижі). Зустрічається і інша важка хромосомна аберація типу делецій: синдром 4 р, синдром 13 q — втрата довгого плеча (q) 13-й хромосоми, синдром 18 р і 18 q і ін. У людини спостерігаються й інші форми хромосомних мутацій. Слід зауважити, що якщо хромосомні перебудови виникають в аутосомах в результаті кросинговеру, вони проявляються у чоловіків і жінок, а перебудови статевих хромосом зустрічаються тільки у жінок (оскільки кон’югують під час мейозу тільки Х-хромосоми).
Транслокації можуть бути незбалансованими, що приводить до патологічних проявів у їх носіїв, і збалансованими, які фенотипічно не виявляються. В потомстві носія (частіше жінки, оскільки вона дає своєму нащадку звичайно тільки одну клітину, запліднену одним з безлічі сперматозоїдів) можливі аномалії.
Геномні мутації
Геномні мутації, пов’язані з втратою або збільшенням пари аутосом або статевих хромосом до трьох, чотирьох і більше, називаються анеуплоідією.
Причина анеуплодії — в анафазі І мейозу гомологічні хромосоми однієї або декількох пар не розійдуться, направляються до одного полюса і утворюють гамети зі змінами кількості хромосом.
Гетероплоїдія за статевими хромосомами
Порушення хромосомних комплексів — гетероплоїдія — в переважній більшості випадків приводить до важких захворювань.
Гетероплоїдія може бути двох типів: моносомія, коли хромосомний комплекс зменшений (2 п – 1), і поліплоїдія, коли число окремих хромосом збільшене, наприклад трисомія (2 п + 1).
Особливо важкі моносомії. Вважають, що близько 20% випадків моносомії закінчуються летально ще на перших днях ембріонального розвитку або приводять до загибелі зародка на пізніших стадіях (спонтанно). Зустрічаються моносомії і серед новонароджених.
У період дозрівання гамет спостерігаються випадки нерозходження статевих хромосом (в І, ІІ або обох поділах дозрівання). У цих випадках гамети несуть не 22 аутосоми + 1 статеву хромосому (X або Y), а спостерігається більше або менше порушення нормальної парності хромосом.
Наведемо короткий опис деяких з цих важких порушень.
Трисомія X (рис. 5), тобто збільшення числа Х-хромосом до трьох (47 хромосом = 44+XXX), проявляється, як правило, у розумовій відсталості, недорозвиненні яєчників і матки у жінок. Проте відомі випадки плодючості таких жінок. В потомстві їх буває значно більше хлопчиків, ніж дівчаток, і серед дітей спостерігається часто порушення хромосомних наборів. В літературі описані жінки більш ніж з трьома Х-хромосомами. Всі жінки з полісомією відрізняються від норми розладом психомоторних функцій.
Для жінок з моносомією — синдром Шерешевського-Тернера ( 45 хромосом = 44+ ХО) характерні патологічні зміни статури (малий ріст, коротка шия, воронкоподібна грудина), порушення статевої системи (безплідність внаслідок недорозвинення яєчників) і будови вторинних статевих ознак (рис. 6). У хворих встановлено стеноз аорти і злиття хребців, вони розумово обмежені. Зустрічається таке захворювання приблизно в однієї з 5 тисяч новонароджених дівчаток. Моносомія X залежить виключно від батька (порушення сперматогенезу). Збільшення числа Х-хромосом у чоловіків більш ніж на одну викликає синдром Кляйнфельтера, який спостерігається в одного з 400–600 новонароджених хлопчиків.
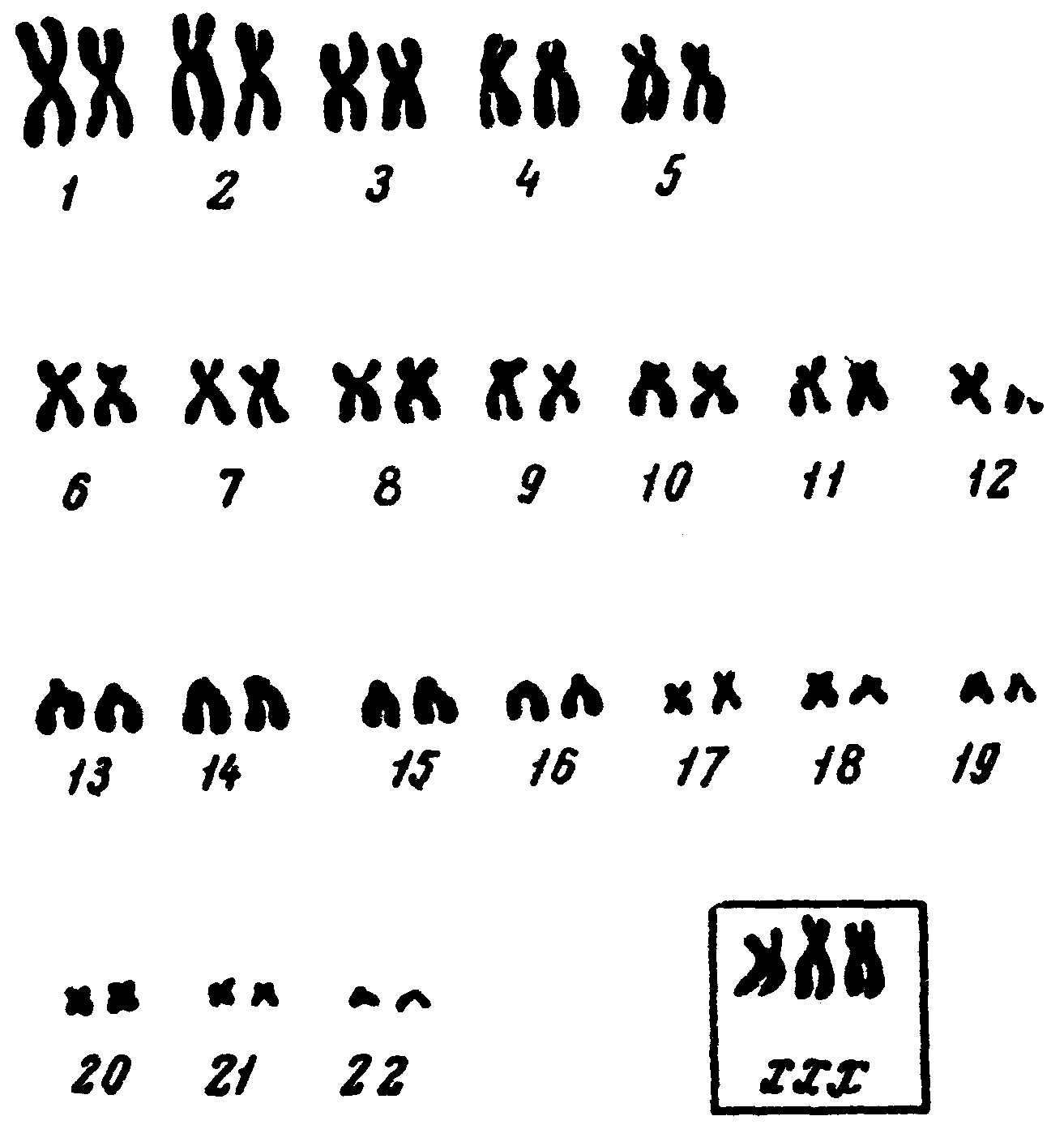
Рис. 5. Каріограма людини при трисомії Х.

Рис. 6. Каріограма при моносомії Х (синдром Шерешевського-Тернера).
У людей з хворобою Кляйнфельтера (XXY, XXXY, XXXXY, XXXXXY) відбувається порушення розвитку і активності статевих залоз, спостерігається євнухоїдизм: більш вузькі, ніж таз, плечі, обволосіння і відкладення жиру на тілі по жіночому типу, довгі в порівнянні з тулубом руки і ноги, високий ріст. Ці ознаки в поєднанні з деякою психічною відсталістю (більш виражена у хворих з великим числом Х-хромосом) виявляються у відносно нормального хлопчика, починаючи з моменту статевого дозрівання. Плодючість знижена. В потомстві зустрічаються порушення числа хромосом.
Іншим варіантом синдрому Кляйнфельтера є полісомія по Y-хромосомі. Описані випадки полісомії XYY (47 хромосом) —фенотипічно це нормальні чоловіки високого росту, діти яких іноді виявляються гетероплоїдами.
Існують дані, що індивідууми, в каріотипі яких виявляється зайва Y-хромосома, дуже жорстокі, агресивні та іноді соціально небезпечні.
Дуже рідко серед хворих з полісомією спостерігаються індивідууми з набором XYYY або навіть XYYYY.
Окрім описаних випадків, зустрічається мозаїцизм по статевих хромосомах, коли одні клітини містять XX-, а інші XY-хромосоми. Таке явище приводить до гермафродитизму.
Гетероплоїдія по аутосомах

| Рис. 8. Каріограма при синдромі Дауна. |
Діагноз часто можна встановити за зовнішніми фенотипічними проявами навіть в новонароджених. Своєрідні форма голови і риси обличчя, монголоїдизм (скошений розріз вузьких очей з епікантусом — нависаючою складкою над верхньою повікою), маленький ніс з широким плоским переніссям), деформовані, частіше невеликі вушні раковини, напіввідкритий рот, недорозвинена верхня і виступаюча нижня щелепі, плоска потилиця, порушення моторики і фізичного розвитку, низький ріст.

| Рис. 7. Каріограма при синдромі Кляйнфельтера. |
Встановлено, що це захворювання може виникати в результаті транслокації зайвої третьої 21-й хромосоми на 15-у, 22-у, 20-у, 4-у, 2-у. Це обставина значно ускладнює цитогенетичну діагностику хвороби. При цьому синдромі зустрічається і мозаїчність (1-2 % від числа всіх хворих), коли одна частина клітин містить нормальний каріотип, а інша — трисомії по 21-й хромосомі. Залежно від співвідношення нормальних і аномальних клітин такі індивідууми можуть займати проміжне положення між здоровими і хворими.
У великих містах є спеціальні інтернати для хворих дітей. Проте батьки рідко віддають дітей з хворобою Дауна на виховання, оскільки ці діти звичайно дуже емоційні і прив’язуються до своїх близьких, співчувають горю, поганому настрою і безмірно радіють радості батьків.
Існує прямий зв’язок між ризиком народження дітей з синдромом Дауна і віком матері.
| Вік матері | Ризик народження дитини з синдромом Дауна |
| 1 на 2000 1 на 290 1 на 40 |
Відомі й інші гетероплоїдії по аутосомам, пов’язані з серйозними відхиленнями від норми. Так, наприклад, синдром Патау, викликаний гетероплоїдією по одній з хромосом групи D (13–15-а), який виявляється в множинних вадах головного мозку (недорозвинення лобових часток, мозочка, третього шлуночка), серцево-судинної системи, нирок і ін., що приводить до ранньої смерті (на 3-4-у місяці життя). Частіше синдром Патау виявляється у дівчаток (1 на 4000 новонароджених). Синдром Едвардса — трисомія 18-й хромосоми (група Е) викликає множинні дефекти життєво важливих органів (головного мозку — зміни клітин кори великих півкуль, атрофія клітин червоного ядра і мозочка; серця, легень, нирок). До одного місяця доживають 70% хворих, ще 7% доживає тільки до року. До 10 років доживає лише 1 % дітей. Як і при синдромі Патау, з трисомією 18 народжуються частіше дівчатка. Трисомія крупних хромосом завжди летальна.
ЗНАЧЕННЯ МЕДИЧНОЇ ГЕНЕТИКИ
Генетика людини, швидко розвиваючись в останні десятиліття, дала відповіді на багато з питань, що давно цікавили людей: від чого залежить стать дитини? Чому діти схожі на батьків? Які ознаки і захворювання успадковуються, а які — ні, чому люди не такі схожі один на одного, чому шкідливі близькоспоріднені шлюби?
Інтерес до генетики людини зумовлений декількома причинами. По-перше, це природне прагнення людини пізнати самого себе. По-друге, після того, як було переможено багато інфекційних хвороб — чума, холера, віспа і ін., — збільшилася відносна частка спадкових хвороб. По-третє, після того, як зрозуміли природу мутацій та їх значення в спадковості, стало ясно, що мутації можуть бути викликані чинниками зовнішнього середовища, на які раніше не звертали належної уваги. Почалося інтенсивне вивчення впливу на спадковість випромінювань і хімічних мутагенів. З кожним роком в побуті, сільському господарстві, харчовій, косметичній, фармакологічній промисловості та інших областях діяльності застосовується все більше хімічних сполук, серед яких використовується немало мутагенів.
Використання генетики в медичній практиці вже сьогодні приносить відчутну користь людству. За допомогою генетики в співпраці з педіатрією, терапією та іншими галузями медицини розвінчаний міф про фатальність і невиліковність спадкових хвороб. Знаючи їх характер, можна вилікувати або запобігти ряду спадкових патологій.
Вже сьогодні у всьому світі (і в нашій країні) велику користь приносять медико-генетичні консультації. Число таких консультацій зростає. За порадою в медико-генетичну консультацію можуть звертатися подружні пари перед прийняттям рішення завести дитину. Звичайно, ніхто не зможе напевно визначити, яким буде нащадок даної конкретної пари. Лікар-генетик може тільки визначити вірогідність, ступінь ризику прояву тієї або іншої спадкової аномалії. Точність оцінки залежить від можливості аналізу захворювання, родоводу сімей і середнього вираження відповідної ознаки в різних сім’ях.
Якщо дитина вже народилась, то консультування може не тільки допомогти в проведенні лікування, але і дати рекомендацію, слід чи не слід народжувати наступну дитину.
Спадкові хвороби та їх причини
Спадкові хвороби можуть бути викликані порушеннями в окремих генах, хромосомах або хромосомних наборах.
Хромосомні хвороби виникають при зміні структури хромосом: подвоєнні або випаданні ділянки хромосоми, повороті ділянки хромосоми на 180°, переміщенні ділянки хромосоми на негомологічну хромосому.
Примітка. Студентам пропонується самостійно сформулювати причини виникнення спадкових захворювань та зростання їх чисельності в останні роки.
Лікування спадкових хвороб
Ефективних засобів лікування спадкових хвороб поки не існує. Проте існують методи лікування, що полегшують стан хворих і поліпшуючих їх самопочуття. Вони засновані головним чином на компенсації дефектів метаболізму, обумовлених порушеннями в геномі.
При спадкових аномаліях обміну речовин хворому вводять ферменти, що не утворюються в організмі, або виключають з харчового раціону продукти, які не засвоюються організмом через відсутність необхідних ферментів.
При цукровому діабеті в організм вводять інсулін. Це дозволяє хворому діабетом нормально харчуватися, проте не усуває причини хвороби.
Чи можна попередити спадкові хвороби? Поки це лише мрія. Проте рання діагностика дозволяє або уникнути народження хворої дитини, або своєчасно почати лікування, що у багатьох випадках дає позитивні результати. Так, наприклад, при ранньому лікуванні синдрому Дауна 44% хворих доживають до віку 60 років, у багатьох випадках ведучи практично нормальний спосіб життя.
Для ранньої діагностики застосовують різні методи. Зазвичай, якщо стандартні методи обстеження дають підстави припускати спадкові порушення у ембріона, застосовують метод амніоцентеза - аналізу клітин ембріона, що завжди присутні в навколоплідній рідині.
Питання і задачі для самоперевірки:
1. У чому полягають особливості вивчення генетики людини?
2. Як успадковуються зчеплені з аутосомами ознаки людини?
3. У чому основні відмінності аутосомно-домінантного і аутосомно-рецесивного типів успадкування?
4. Батько родини (мати якого була гh–, I група) Rh+, III група, була гh–, I група. Які можливі варіанти груп крові і резус-фактора у потомства?
5. Чому резус-конфлікт виникає при шлюбі «резуспозитивного» чоловіка і «резуснегативної» жінки, а не навпаки?
6. Батько має I групу крові, мати — III групу. Яка група крові може бути у дітей?
7. У суді слухається справа з приводу стягнення аліментів. Мати має I групу крові, дитина — II групу. Чи може бути батьком дитини чоловік з III групою крові? Яка група крові можлива для батька?
8. Батько — ахондропластичний карлик, здатний сприймати смак фенілтіокарбаміду. Мати нормального росту, має «шерстисте» волосся; смак фенілтіокарбаміду не сприймає. Всі нащадки від цього шлюбу мають нормальний ріст, не шерстисте волосся, смак фенілтіокарбаміду сприймають. Визначити генотипи всіх членів сім’ї.
Контрольні питання до ТЕМИ
СПАДКОВІ ЗАХВОРЮВАННЯ ЛЮДИНИ
1. Згрупуйте у два стовпчики захворювання людини, інформація про які локалізована в аутосомах або статевих хромосомах:
а) синдром Дауна;
б) синдром котячого крику;
в) гемофілія;
г) синдром Кляйнфельтера.
2. Які захворювання локалізовані на Y-хромосомі?
а) перетинки між пальцями;
б) волохаті вуха;
в) дальтонізм;
г) пігментна ксеродерма.
3. Перерахуйте симптоми альбінізму.
4. Хто хворіє на гемофілію — чоловіки чи жінки? Чому?