Главная страница Случайная страница
КАТЕГОРИИ:
АвтомобилиАстрономияБиологияГеографияДом и садДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеталлургияМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРелигияРиторикаСоциологияСпортСтроительствоТехнологияТуризмФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Экспериментальные и клинические проявления растормаживания
|
|
Характерным экспериментальным синдромом растормаживания является децеребрационная ригидность. Она вызывается, по Шеррингтону, перерезкой ствола мозга между передним и задним четверохолмием. В этих условиях происходит выпадение тормозных влияний со стороны супраспинальных структур и особенно красных ядер и проявляются возбуждающие тонические влияния вестибулярных ядер Дейтерса на мотонейроны спинного мозга, особенно гамма-мотонейроны, которые в норме находятся под тормозным контролем со стороны красных ядер. Перерыв (например, путем перерезки задних корешков) расторможенной, патологически усиленной гамма-петли на уровне спинного мозга ведет к исчезновению ригидности соответствующих мышц. Поэтому данный вид децеребраци-онной ригидности называют также гамма-ригидностью (Р. Гранит).
При выпадении тормозных влияний растормаживаются и гиперактивируются прежде всего те нейроны, которые в норме находятся в состоянии тонического возбуждения. У человека и многих животных такими нейронами являются нейроны мышц, выполняющих антигравитационную функцию. Вследствие этого у децереб-рированной кошки голова запрокидывается вверх, передние и задние лапы вытягиваются, хвост поднимается и т.п. У человека при выпадении моторных корковых влияний (например, после геморрагического инсульта) возникает спастическая флексорная установка верхней и эк-стензорная установка нижней конечностей (поза Вернике - Манна).
Часть III. ПАТОФИЗИОЛОГИЯ ОРГАНОВ И СИСТЕМ
Целый ряд патологических рефлексов возникает в условиях выпадения влияний со стороны коры и подкорковых образований; эти рефлексы являются результатом растормаживания центров спинного или продолговатого мозга. Они представляют собой гиперболизированные неконтролируемые реакции, которые были нормальными в раннем постнатальном периоде и затем подавлены при развитии регулирующих влияний со стороны высших отделов ЦНС. К ним относятся рефлекс Бабинского (растопыривание пальцев ноги вместо их сгибания при раздражении подошвы), хватательный, сосательный и другие рефлексы.
При полном перерыве спинного мозга могут проявляться заложенные генетически и подавленные с возрастом спинальные автоматизмы в виде сравнительно координированных сгиба-гельно-разгибательных движений конечностей. Если растормаживаются и гиперактивируются тормозные нейроны, то возникает патологически усиленный тормозной эффект, который может проявиться в виде угнетения и выпадения функции.
20.2.3. Денервационный синдром
Денервационный синдром представляет собой комплекс изменений, возникающих в постсинап-тических нейронах, органах и тканях после выпадения нервных влияний на эти структуры. Денервированная структура (мышца, нейрон) приобретает повышенную чувствительность к физиологически активным веществам (закон Кеннона - Розенблюта). Основным проявлением денервационного синдрома в мышце является исчезновение концевой пластинки - зоны мышечного волокна, где сосредоточен весь его холи-нергический аппарат. Вместо нее появляются новые АХ-рецепторы на всем протяжении мышечного волокна, и в связи с этим происходит повышение общей чувствительности к АХ всего волокна (А.Г. Гинецинский, Н.М. Ашмарина). Этот эффект связан главным образом с выпадением трофических влияний с нерва. Другой характерный признак - фибриллярные подергивания денервированной мышцы. Этот эффект отражает реакцию мышечных денервирован-ных волокон на поступающий к ним из разных сторонних источников АХ. Близок к этим проявлениям эффект Вюльпиана - Гейденгайна -тономоторное сокращение денервированной мышцы при раздражении нерва, выделяющего
АХ, что в норме вызывает лишь сосудистые реакции.
При денервации в мышце и других тканях появляются свойства, присущие ранним, в частности, эмбриональным стадиям развития. Это явление возникает как результат патологического растормаживания супрессированных в норме генов и других процессов.
20.2.4. Деафферентация
Импульсация, поступающая в нейрон, из какого бы источника она ни исходила, является для нейрона афферентной. Выключение этой афферентации представляет собой деафферента-цию нейрона. Последняя может быть обусловлена либо выпадением поступающей импульсации (при перерыве нервных путей, нарушении выделения нейромедиаторов пресинаптическими окончаниями), либо блокадой воспринимающих рецепторов на постсинаптическом нейроне (при действии токсинов, фармакологических средств и др.).
Многие явления при деафферентации нейрона представляют собой выражение денервационного синдрома. Полной деафферентации нейрона не происходит, так как нейроны ЦНС обладают огромным количеством афферентных входов. Тем не менее и при частичной деафферентации возникает повышение возбудимости нейрона или его отдельных участков и нарушение тормозных механизмов. В силу этого при деафферентации группа нейронов может приобрести свойства генератора.
В клинике под феноменом деафферентации имеют в виду синдромы, возникающие при выпадении афферентной стимуляции с периферии. Эти синдромы можно воспроизвести в эксперименте путем перерезки соответствующих задних корешков. Движения конечности, иннервируе-мой деафферентированными таким образом сегментами спинного мозга, становятся размашистыми, плохо координированными. Кроме того, такая конечность способна осуществлять спонтанные движения в такт с дыханием (феномен Орбели - Кунстман), что обусловлено рас-тормаживанием и повышением возбудимости деафферентированных нейронов спинного мозга.
20.2.5. Спинальный шок
Спинальный шок возникает после перерыва спинного мозга и представляет собой глубокое,
 но обратимое угнетение двигательных и вегетативных рефлексов, осуществляющихся ниже перерыва. Угнетение рефлексов связано с выпадением активирующей стимуляции со стороны головного мозга. У лягушек, у которых зависимость спинного мозга от головного значительно меньшая, чем у высших животных, спинальный шок длится несколько минут, у человекообразных обезьян и человека - несколько месяцев.
но обратимое угнетение двигательных и вегетативных рефлексов, осуществляющихся ниже перерыва. Угнетение рефлексов связано с выпадением активирующей стимуляции со стороны головного мозга. У лягушек, у которых зависимость спинного мозга от головного значительно меньшая, чем у высших животных, спинальный шок длится несколько минут, у человекообразных обезьян и человека - несколько месяцев.
У человека полная арефлексия после перерыва спинного мозга является начальной стадией полной параплегии. В дальнейшем происходит постепенное восстановление двигательных и вегетативных рефлексов. Вначале появляются сги-бательные рефлексы пальцев, имеющие характер патологических рефлексов (рефлекс Бабинс-кого и др.), после этого осуществляются более значительные и затем генерализованные спи-нальные рефлексы и движения типа спиналь-ных автоматизмов.
20.2.6. Нарушение нервной трофики. Нейродистрофический процесс
Трофика клетки и дистрофический процесс.
Трофика клетки - комплекс процессов, обеспечивающих ее жизнедеятельность и поддержание генетически заложенных свойств. Расстройство трофики представляет собой дистрофию, развивающиеся дистрофические изменения составляют дистрофический процесс.
Этиологические факторы, вызывающие дистрофию клетки, могут быть различной природы. Развитие же дистрофического процесса носит стандартный характер. Эта особенность связана с тем, что внутриклеточные процессы протекают в виде цепных реакций, имеющих определенную последовательность. Поэтому дистрофический процесс в клетке относится к числу типовых внутриклеточных патологических процессов (Г. Н. Крыжановский).
Дистрофический процесс может развиваться и на тканевом, и на органном уровне, и на уровне организма. В той или иной форме он возникает при всех видах патологии, играя роль вторичного или параллельно развивающегося неспецифического эндогенного повреждения, входящего в комплекс патогенетических механизмов данной формы патологии.
Нейродистрофический процесс. Это развивающееся нарушение трофики, которое обусловлено выпадением или изменением нервных влияний. Оно может возникать как в периферичес-
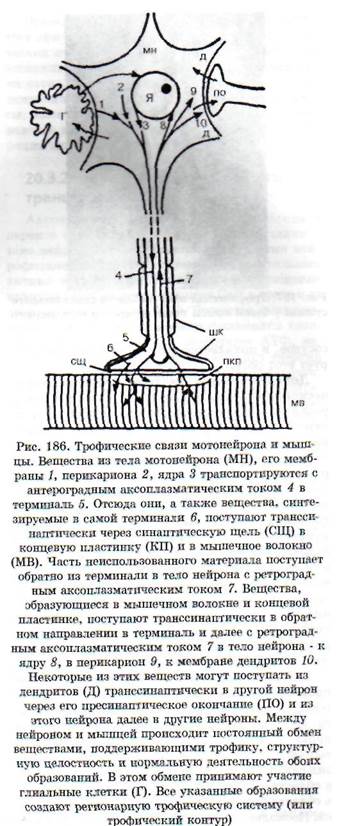
ких тканях, так и в самой нервной системе. Выпадение нервных влияний заключается: 1) в прекращении функциональной стимуляции ин-нервируемой структуры в связи с нарушением выделения или действия нейромедиатора; 2) в нарушении секреции или действия комедиато-ров - веществ, которые выделяются вместе с ней-ромедиаторами и играют роль нейромодулято-ров, обеспечивающих регуляцию рецепторных, мембранных и метаболических процессов; 3) в нарушении выделения и действия трофогенов. Трофогены (трофины) - вещества различной, преимущественно белковой природы, осуществляющие собственно трофические эффекты поддержания жизнедеятельности и генетически заложенных свойств клетки. Источником трофогенов являются: 1) нейроны, из которых трофогены поступают с ортоградным аксоплазматичес-ким током в клетки-реципиенты (другие нейроны или иннервируемые ткани на периферии); 2) клетки периферических тканей, из которых трофогены поступают по нервам с ретроградным аксоплазматическим током в нейроны (рис. 186); 3) глиальные и шванновские клетки, которые обмениваются с нейронами и их отростками трофическими веществами. Вещества, играющие роль трофогенов, образуются также из сывороточных и иммунных белков. Трофическое воздействие могут оказывать некоторые гормоны. В регуляции трофических процессов принимают участие пептиды, ганглиозиды, некоторые нейромедиаторы.
К нормотрофогенам относятся различного рода белки, способствующие росту, дифферен-цировке и выживанию нейронов и соматических клеток, сохранению их структурного гомеостаза (например, фактор роста нервов).
В условиях патологии в нервной системе возникают трофические вещества, индуцирующие устойчивые патологические изменения клеток-реципиентов (патотрофогены, по Г. Н. Крыжа-новскому). Такие вещества синтезируются, например, в эпилептических нейронах - поступая с аксоплазматическим током в другие нейроны, они могут индуцировать у этих нейронов-реципиентов эпилептические свойства. Патотрофогены могут распространяться по нервной системе, как по трофической сети, что является одним из механизмов распространения патологического процесса. Патотрофогены образуются и в других тканях.
Часть III. ПАТОФИЗИОЛОГИЯ ОРГАНОВ И СИСТЕМ
|
Дистрофический процесс в денервированной мышце. Синтезируемые в теле нейрона и транспортируемые в терминаль с аксоплазматическим током вещества, как и вещества, образующиеся в терминал и, выделяются нервным окончанием и поступают в мышечные волокна (см. рис. 186), выполняя функцию трофогенов. Эффекты ней-ротрофогенов видны из опытов с перерезкой двигательного нерва - чем выше произведена перерезка, т.е. чем больше сохранилось трофогенов в периферическом отрезке нерва, тем позднее наступает денервационный синдром. Нейрон вместе с иннервируемой им структурой (например, мышечным волокном) образует регионарный трофический контур, или регионарную трофическую систему (рис. 186). Если осуществить перекрестную реиннервацию мышц с разными исходными структурно-функциональными характеристиками (реиннервация «медленных» мышц волокнами от нейронов, иннервировавших «быстрые» мышцы, и наоборот), то реиннерви-рованная мышца приобретает в значительной мере новые динамические характеристики: «медленная» становится «быстрой», «быстрая» - «медленной».
В денервированной мышечном волокне возникают новые трофогены, которые активируют разрастание нервных волокон (sprouting). Указанные явления исчезают после реиннервации.
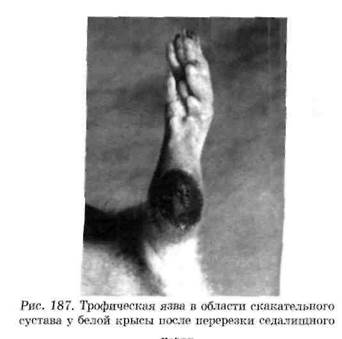
Нейродистрофический процесс в других тканях. Взаимные трофические влияния существуют между каждой тканью и ее нервным аппаратом. При перерезке афферентных нервов возникают дистрофические изменения кожи. Перерезка седалищного нерва, который является смешанным (чувствительным и двигательным), вызывает образование дистрофической язвы в области скакательного сустава (рис. 187). С течением времени язва может увеличиться в размерах и охватить всю стопу.
Классический опыт Ф. Мажанди (1824), послуживший началом разработки всей проблемы нервной трофики, заключается в перерезке у кролика первой ветви тройничного нерва. В результате такой операции развивается язвенный кератит, вокруг язвы возникает воспаление и в роговицу врастают со стороны лимба сосуды, которые в норме в роговице отсутствуют. Врастание сосудов является выражением патологического растормаживания сосудистых элементов - в дистрофически измененной роговице исчезает фактор, который тормозит в норме рост в нее
|
нерва
сосудов, и появляется фактор, который активирует этот рост.
Дополнительные факторы нейродистрофи- ческого процесса. К факторам, участвующим в развитии нейродистрофического процесса, относятся: сосудистые изменения в тканях, нарушения гемо- и лимфомикроциркуляции, патологическая проницаемость сосудистой стенки, нарушение транспорта в клетку питательных и пластических веществ. Важным патогенетическим звеном является возникновение в дистрофической ткани новых антигенов в результате изменений генетического аппарата и синтеза белка, образуются антитела к тканевым антигенам, возникают аутоиммунный и воспалительный процессы. В указанный комплекс патологических процессов входят также вторичное инфицирование язвы, развитие инфекционных повреждений и воспаления. В целом нейродист-рофические поражения тканей имеют сложный многофакторный патогенез (Н. Н. Зайко).
Генерализованный нейродистрофический процесс. При повреждениях НС могут возникать генерализованные формы нейродистрофического процесса. Одна из них проявляется в виде поражения десен (язвы, афтозный стоматит), выпадения зубов, кровоизлияния в легких, эрозии слизистой и кровоизлияния в желудке (чаще в области привратника), в кишечнике, особенно в области буагиниевой заслонки, в прямой кишке. Поскольку такие изменения возникают срав-
нительно регулярно и могут иметь место при разных хронических нервных повреждениях, они получили название стандартной формы нервной дистрофии (А. Д. Сперанский). Весьма часто указанные изменения возникают при повреждении высших вегетативных центров, в частности гипоталамуса (при травмах, опухолях), в эксперименте при наложении стеклянного шарика на турецкое седло.
Трофическая иннервация и трофические системы. Все нервы (двигательные, чувствительные, вегетативные, внутрицентральные нервные связи) осуществляют непосредственные трофические взаимодействия с иннервируемым ими субстратом (периферические ткани, постсинап-тические нейроны). Поэтому каждый нерв, какую бы функцию он ни выполнял, является одновременно трофическим (А. Д. Сперанский). Наряду с этим существует и специальная иннервация (усиливающие нервы, по И. П. Павлову; адаптационно-трофическая симпатическая нервная система, по Л. А. Орбели), возникшая в процессе эволюционного развития, которая регулирует в соответствии с текущими потребностями метаболизм органа, его энергетические и трофико-пластические процессы, усиливает при необходимости эти процессы и способствует быстрейшему восстановлению трофического и энергетического потенциала органа при его функционировании.
Нервная система в целом, благодаря многочисленным и разнообразным нейрональным связям, представляет собой трофическую сеть. По этой сети распространяются также патогенные факторы эндогенной (патотрофогены, антитела к нервной ткани и нейромедиаторам) и экзогенной (токсины, вирусы) природы. В теснейшем трофическом взаимодействии функционируют все главные интегративные системы организма - нервная, эндокринная, иммунная. Наряду с этим периферические ткани, находясь под трофическим контролем, сами оказывают трофическое влияние на нервную и другие интегративные системы. Таким образом, существует единая трофическая система организма (Г.Н. Кры-жановский). Первичные нарушения в каком-либо звене этой системы влекут за собой изменения и в других звеньях.
Нарушения нервной трофики составляют важное патогенетическое звено болезней нервной системы и нервной регуляции соматических органов, поэтому коррекция трофических измене-
Часть III. ПАТОФИЗИОЛОГИЯ ОРГАНОВ И СИСТЕМ
ний является необходимой частью комплексной патогенетической терапии.
20.3. ПАТОЛОГИЯ НЕЙРОНА
20.3.1. Нарушение проведения возбуждения
Распространение возбуждения по нервному волокну обеспечивается последовательным сочетанием одних и тех же процессов: деполяризацией участка мембраны волокна > входом в этом участке Na* > деполяризацией соседнего участка мембраны > входом в этом участке Na+ и т.д.
При недостаточном входе Na* нарушается генерация распространяющегося потенциала действия и проведение прекращается. Такой эффект имеет место при блокаде Ыа+-каналов местными анестетиками (новокаин, лидокаин и др.) и рядом других химических агентов. Специфическим блокатором Na^-каналов является тетродотоксин - яд, вырабатывающийся во внутренних органах рыбы фугу. Блокирование проведения возбуждения вызывают также вещества, нарушающие процесс реполяризации мембраны, связанный с закрыванием Na'-каналов. К ним относятся инсектициды (например, ДДТ), вератридин, аконитин, батрахотоксин и др.
Исходная разность концентрации ионов Na* и К* по обе стороны мембраны (Na+в 10-15 раз больше снаружи, К+ в 50-70 раз больше внутри), необходимая для генерации потенциала действия, восстанавливается и поддерживается активным транспортом ионов Na+-, К+-насосом. Он выкачивает наружу Na', поступивший внутрь (в цитоплазму) во время возбуждения, в обмен на наружный К+, который вышел наружу во время возбуждения. Деятельность насоса, роль которого выполняет встроенная в мембрану Na*-, K*-АТФаза, обеспечивается энергией, высвобождающейся при расщеплении АТФ. Дефицит энергии ведет к нарушению работы насоса, что обусловливает неспособность мембраны генерировать потенциал действия и проводить возбуждение. Такой эффект вызывают разобщители окислительного фосфорилирования (например, динит-рофенол) и другие метаболические яды, а также ишемия и длительное охлаждение участка нерва. Ингибируют насос и, как следствие этого, нарушают проводимость сердечные гликозиды (например, уабаин, строфантин) при их применении в относительно больших дозах.
Проведение возбуждения по аксону нарушается при различных видах патологии периферических нервов и нервных волокон в ЦНС - при воспалительных процессах, Рубцовых изменениях нерва, при сдавлении нервных волокон, при демиелинизации волокон (аллергические процессы, рассеянный склероз), при ожогах и др. Проведение возбуждения прекращается при дегенерации аксона.
20.3.2. Нарушение аксонального
транспорта
Аксональный транспорт из тела нейрона в нервное окончание и из нервного окончания в тело нейрона осуществляется при участии ней-рофиламентов, микротрубочек и контрактильных актино- и миозиноподобных белков, сокращение которых зависит от содержания Саг+ в среде и от энергии расщепления АТФ. Вещества, разрушающие микротрубочки и нейрофиламенты (кол-хицин.винбластин и др.), недостаток АТФ, метаболические яды, создающие дефицит энергии (динитрофенол, цианиды), нарушают аксоток. Аксональный транспорт страдает при дегенерации аксона, вызываемой недостатком витамина В6 и витамина В,) (болезнь бери-бери), промышленными ядами (например, акриламидом, гек-сахлорофосом), солями тяжелых металлов (например, свинца), фармакологическими препаратами (например, дисульфирамом), алкоголем, при диабете, при сдавлении нервов и при дистрофических повреждениях нейрона. При перерыве аксона возникает уоллеровская дегенерация (распад) его периферической части и ретроградная дегенерация центральной части. Эти процессы связаны с нарушением трофики в обеих частях аксона.
Расстройства аксонального транспорта трофо-генов и веществ, необходимых для образования и выделения медиаторов нервным окончанием, обусловливают развитие дистрофических изменений нейронов и иннервируемых тканей и нарушение синаптических процессов. Распространение с аксональным транспортом патотрофоге-нов, антител к нервной ткани и к нейромедиато-рам приводит к вовлечению в патологический процесс нейронов в соответствующих отделах ЦНС.
20.3.3. Патология дендритов
Дендриты и их шипики являются самыми
ранимыми структурами нейрона. При старении шипики и ветви дендритов редуцируются, при некоторых дегенеративных и атрофических заболеваниях мозга (старческое слабоумие, болезнь Альцгеймера) они не выявляются. Дендро-ши-пиковый аппарат страдает при гипоксии, ишемии, сотрясении мозга, стрессорных и невроти-зирующих воздействиях. Патология дендритов связана также с нарушением их микротрубочек, которые исчезают при действии различных патогенных агентов.
20.3.4. Патология нейрональных мембран
Повреждения как клеточной (цитоплазмати-ческой), так и внутриклеточных мембран возникают при различных патогенных воздействиях, и сами являются причиной дальнейшего развития патологии нейрона.
Усиленное перекисное окисление липидов (ПОЛ) нейрональных мембран оказывает влияние не только на мембранные, но и на другие внутриклеточные процессы.
Практически нет патологического процесса в нервной системе, при котором не возникало бы усиленного ПОЛ. Оно имеет место при эпилепсии, эндогенных психозах (например, шизофрении, маниакально-депрессивном синдроме), при неврозах, различного рода стрессах и повреждениях, при ишемии, хронической гипоксии, функциональных перегрузках нейронов и пр. С ним связана дальнейшая гиперактивация нейронов.
Вследствие увеличения проницаемости мембран происходит выход из нейрона различных веществ, в том числе антигенов, которые вызывают образование антинейрональных антител, что приводит к развитию аутоиммунного процесса. Нарушение барьерных свойств мембран обусловливает возрастание тока ионов Са2* и Na' в нейрон и К+ - из нейрона; эти процессы в сочетании с недостаточностью энергозависимых Na+-, K+- и Са2+-насосов (их деятельность изменяется также под влиянием усиленного ПОЛ) приводят к частичной деполяризации мембраны. Увеличенный вход Са2+ не только вызывает гиперактивацию нейрона, но и при чрезмерном его содержании в клетке ведет к патологическим изменениям метаболизма и внутриклеточным повреждениям. Весь указанный комплекс процессов, если он не подавляется и не компенсируется, обусловливает гибель нейрона.
Нормализация ПОЛ и стабилизация нейрональных мембран должны быть частью комплексной патогенетической терапии различных форм патологии НС.
20.3.5. Энергетический дефицит
Потребность нейронов в энергообеспечении -самая высокая из всех клеток организма, и нарушение энергообеспечения является одной из распространенных причин патологии нейрона. Энергетический дефицит может быть первичным - при действии метаболических ядов (например, динитрофенола, цианидов), либо вторичным - при различных повреждениях, нарушениях кровообращения, шоке, отеке, общих судорогах, усиленной функциональной нагрузке и др. Дефицит энергии относится к разряду типовых внутриклеточных патологических процессов.
Главными условиями развития энергетического дефицита являются недостаток кислорода и значительное повреждение митохондрий, в которых синтезируется основной носитель энергии - АТФ. Причиной дефицита энергии может быть также недостаток субстрата окисления, в частности глюкозы, которая является для мозга основным субстратом окисления. Нейроны коры не имеют запасов глюкозы и потребляют ее непосредственно из крови (глюкоза свободно проходит ГЭБ), поэтому они особенно чувствительны к гипогликемии. Мозг потребляет около 20% от всей находящейся в крови глюкозы. Инсули-новые шоки, применяемые для лечения некоторых психозов, связаны с глубокой гипогликемией и протекают с потерей сознания и нередко с судорогами. При ряде патологических состояний (травматический шок, кровопотеря) мозг может дольше обеспечиваться кислородом и глюкозой благодаря перераспределению крови и уменьшению их потребления другими тканями. Для быстрейшего восстановления деятельности мозга после общих судорог необходим достаточно высокий уровень глюкозы в крови. Энергетический дефицит усугубляется нарушением цикла Кребса.
При глубоком нарушении окислительного фосфорилирования и синтеза макроэргов источником энергии становится анаэробный гликолиз. Он имеет характер компенсаторного механизма, однако его эффект не может восполнить дефицит энергии, а нарастающее увеличение содержания молочной кислоты в мозге оказывает от-
рицательное влияние на деятельность нейронов и усугубляет отек мозга.
20.3.6. Эффекты ишемии и гипоксии
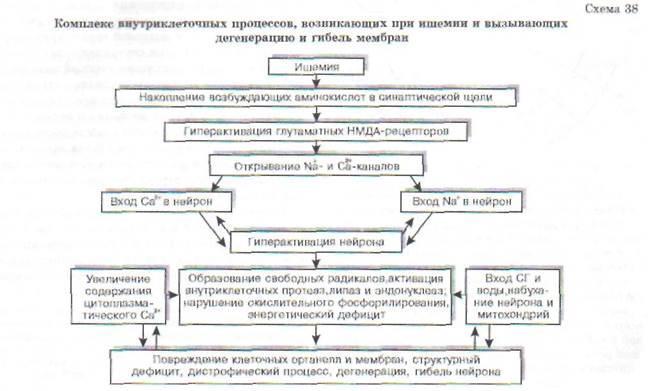
В связи с высокой потребностью в энергии нейроны ЦНС нуждаются в значительном кислородном обеспечении. Нейрон коры головного мозга потребляет 250-450 мкл 02/мин (для сравнения - глиоцит и гепатоцит потребляют до 60 мкл 02). Снижение потребления кислорода мозгом всего лишь на 20% может вызвать потерю сознания у человека. Исчезновение импульсной активности нейронов возникает уже в первые десятки секунд ишемии мозга. Через 5-6 мин после начала асфиксии наступает глубокое и нередко необратимое нарушение деятельности мозга. Гибель нейрона при ишемии является результатом осуществления комплекса взаимосвязанных внутриклеточных процессов (схема 38).
При аноксии головного мозга в первую очередь страдает кора. Гибель всего мозга означает «мозговую смерть», которая проявляется в полном исчезновении биоэлектрической активности. При наступлении «мозговой смерти» согласно законодательству можно брать у погибшего
внутренние органы для пересадки - они еще функционируют, так как более резистентны к аноксии, чем ЦНС. Филогенетически более старые структуры ЦНС (спинной мозг, ствол головного мозга) менее чувствительны к асфиксии, чем молодые (подкорка и особенно кора). Поэтому при запоздалом оживлении организма может наступить декортикация, «бескорковый» организм может существовать на искусственном дыхании.
Весьма чувствительны к аноксии тормозные механизмы. Одним из следствий этого является растормаживание неповрежденных структур ЦНС. На ранних стадиях ишемии, когда нейроны мозга еще способны давать реакцию, они могут гиперактивироваться. На поздних стадиях ишемии гиперактивация нейронов сменяется их инактивацией.
С входом Na+ в нейрон связана первая, острая фаза поражения нейрона. Возрастание концентрации Na+ в цитозоле нейрона приводит к повышению осмолярности, что обусловливает вход воды в нейрон и его набухание. В дальнейшем повышение осмолярности нейрона связано также с накоплением в нем Са2', молочной кислоты, неорганического фосфора. С входом Са2+в
Глава 20 / ПАТОФИЗИОЛОГИЯ НЕРВНОЙ СИСТЕМЫ
|
нейрон связана вторая фаза повреждения нейрона. Возросший вход Са24 в нейрон обусловлен активацией глютаматных рецепторов в связи с усиленным выделением глютамата нервными окончаниями при ишемии. Антагонисты глютаматных рецепторов и антагонисты Са2т (блока-торы Са2+-каналов) способны предотвратить ише-мическую дегенерацию нейронов и оказать лечебный эффект.
Повреждение нейрона происходит не только во время ишемии, но и после нее. Эти повреждения связаны с реперфузией мозга и возобновлением циркуляции крови, и именно они могут представлять главную опасность. Большую роль реперфузионных постишемических повреждениях играют: новая волна поступления Ca2f в нейрон, перекисное окисление липидов и процессы свободнорадикального окисления, усиленные в связи с действием поступающего кислорода -
возрастание содержания молочной кислоты в связи с поступлением глюкозы в условиях нарушения окислительного фосфорилирования и в связи с возросшим анаэробным гликолизом; происходит отек мозга за счет поступления воды из крови при возобновлении циркуляции.
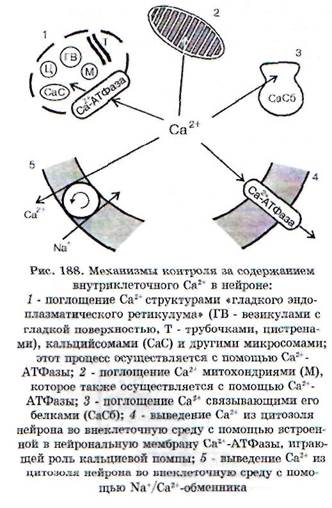
Чрезмерное содержание Са2' в нейроне, с чем связано возникновение ишемической и постише-мической дегенерации нейрона, обусловлено не только усиленным входом Са2+, но и нарушением механизмов его внутриклеточного гомеоста-за (рис. 188). В сложный комплекс Са2+-индуци-руемых внутриклеточных повреждений входят: альтерация внутриклеточных белков, усиленный фосфолипазный гидролиз и протеолиз, разрушение внутриклеточных структур, повреждение цитоплазматической и внутриклеточных мембран, набухание нейронов, нарушение деятельности генома. При критическом возрастании интенсивности этих процессов происходят необратимые повреждения и гибель нейрона, возникает так называемая «кальциевая смерть»*.
На поздних стадиях патологического процесса, вызванного ишемией мозга, а также при хро-низации процесса возникает новый комплекс вторичных изменений - дегенеративно-дистрофические процессы, нарушения энзимных и метаболических систем, сосудистые изменения, образование антител к мозговой ткани, аутоиммунная агрессия и др. Они составляют патогенетическую структуру постишемической энцефалопатии, которая может продолжать развиваться (прогредиентное развитие). Эти процессы, а также изменения в других системах и органах с их последствиями имеют место и после реанимации организма, особенно если она была затяжной и поздней. В своей совокупности они составляют патогенетическую структуру постреанимационной болезни (В.А. Неговский).
Гипоксия той или иной степени сопровождает многие (если не все) формы патологии мозга. Являясь типовым и неспецифическим процессом, она, однако, может вносить значительный вклад в его развитие. Вместе с тем умеренная
' Нарушение внутриклеточного гомеостаза Са2' может иметь место не только при ишемии, но и при других формах патологии нервной системы, при чрезмерной и длительной гиперактивации нейрона, особенно в условиях энергетического дефицита, при усиленном действии глютамата и пр. Оно относится к типовым внутриклеточным патологическим процессам.
Часть III. ПАТОФИЗИОЛОГИЯ ОРГАНОВ И СИСТЕМ
гипоксия может стимулировать метаболические и пластические процессы в нейроне, способствовать адаптации и повышению резистентности, повышать трофический и пластический потенциал нейрона, усиливать адаптационные возможности мозга. Дозированная гипоксия применяется для профилактики и лечения ряда заболеваний центральной нервной системы.
20.3.7. Синаптическая стимуляция и повреждение нейронов
Возбуждающая синаптическая стимуляция может играть важную роль в развитии патологии нейрона. Усиленная и длительная синаптическая стимуляция сама по себе вызывает функциональное перенапряжение нейрона, его стресс, который может завершиться дегенерацией внутриклеточных структур. Стрессорные повреждения усиливаются при нарушениях микроциркуляции и мозгового кровообращения, при действии токсических факторов.
Первостепенное значение синаптическая стимуляция имеет при развитии аноксических (ише-мических) повреждений. Культура тканей нейронов становится чувствительной к аноксии лишь после установления синаптических контактов между нейронами. Весьма чувствительны к аноксии нейроны коры и гиппокампа, в которых имеется высокая плотность возбуждающих синаптических входов. Синаптическая стимуляция реализуется через действие возбуждающих аминокислот (глутамат, аспартат, L-гомоцистеинат), причем эти повреждения подобны тем, которые возникают при ишемии и связаны с увеличенным содержанием внутриклеточного Ca2t. Этот эффект известен как нейротоксическое (или цитотоксическое) действие возбуждающих аминокислот. С синаптической гиперактивацией, действием возбуждающих аминокислот и гипоксией связаны повреждение и гибель нейронов при эпилептическом статусе и в постишемичес-ком периоде. При этом к патогенному действию указанных факторов присоединяется энергетический дефицит.
В связи с изложенным становятся понятными благоприятные эффекты (т.е. ослабление си-наптического воздействия) уменьшения функциональной нагрузки, предотвращение дополнительных раздражений, «охранительное», по И.П. Павлову, торможение обратимо поврежденных нейронов.
20.3.8. Нарушение структурного
гомеостаза нейрона
Значительную роль в патологии нейрона играют нарушения внутриклеточного структурного гомеостаза. В норме процессы изнашивания и распада внутриклеточных структур уравновешиваются процессами их обновления и регенерации. Совокупность этих процессов составляет динамический структурный внутриклеточный гомеостаз.
Внутриклеточная регенерация - универсальный биологический механизм, имеющий место во всех клетках организма. Для жизнедеятельности нейрона, который, как высокодифферен-цированная клетка, не способен митотически делиться, этот механизм имеет существенное значение - внутриклеточная регенерация является единственным способом структурного обновления нейронов и поддержания их целостности. К ней относятся синтез белков, образование внутриклеточных органелл, митохондрий, мембранных структур, рецепторов, рост нервных отростков (аксоны, дендриты, дендритные шипики) и
ДР-
Процессы внутриклеточной регенерации требуют высокого энергетического и трофического обеспечения и полноценного метаболизма клетки. При повреждениях нейрона, возникновении энергетического и трофического дефицита, нарушениях деятельности генома страдает внутриклеточная регенерация, падает пластический потенциал клетки, распад внутриклеточных структур не уравновешивается их восстановлением - происходят глубокие нарушения динамического структурного гомеостаза нейрона; при прогрессировании этого процесса нейрон погибает.
20.3.9. Нарушение деятельности
нейрона при изменении процессов
внутриклеточной сигнализации
После восприятия рецептором сигнала (связывания рецептором нейромедиатора, гормона и др.) в нейроне возникает каскад цепных метаболических процессов, обеспечивающих необходимую активность нейрона. Существенную роль в этих процессах играют так называемые усилительные, или пусковые, ферменты и образующиеся под их влиянием вещества-посредники, вторичные мессенджеры.
 Два типа из указанных процессов наиболее изучены: в одном из них (система АЦ-аза -цАМФ) роль пускового усилительного фермента играет аденилатциклаза (АЦ-аза), а роль связанного с ней вторичного мессенджера - циклический аденозинмонофосфат (цАМФ); в другом (система фосфоинозитидов) пусковым ферментом является фосфолипаза С, а в качестве вторичных мессенджеров выступают инозиттрифосфат (ИФ.1) и диацилглицерин (ДАГ). Роль универсального вторичного мессенджера играет Са2*, принимающий участие практически во всех внутриклеточных процессах. Существенным результатом деятельности указанных систем и Са2+ является активация ряда протеинкиназ, которые обеспечивают фосфорилирование и повышение, таким образом, активности различных функциональных белков - мембранных, цитоплазма-тических и ядерных, ионных каналов, с чем связаны осуществление функций нейрона и его жизнедеятельность.
Два типа из указанных процессов наиболее изучены: в одном из них (система АЦ-аза -цАМФ) роль пускового усилительного фермента играет аденилатциклаза (АЦ-аза), а роль связанного с ней вторичного мессенджера - циклический аденозинмонофосфат (цАМФ); в другом (система фосфоинозитидов) пусковым ферментом является фосфолипаза С, а в качестве вторичных мессенджеров выступают инозиттрифосфат (ИФ.1) и диацилглицерин (ДАГ). Роль универсального вторичного мессенджера играет Са2*, принимающий участие практически во всех внутриклеточных процессах. Существенным результатом деятельности указанных систем и Са2+ является активация ряда протеинкиназ, которые обеспечивают фосфорилирование и повышение, таким образом, активности различных функциональных белков - мембранных, цитоплазма-тических и ядерных, ионных каналов, с чем связаны осуществление функций нейрона и его жизнедеятельность.
Совокупность указанных каскадных мембранных и внутриклеточных процессов составляет эндогенную усилительную систему нейрона, которая может обеспечить многократное усиление входного сигнала и возрастание его эффекта на выходе из нейрона. Так, каскад метаболических процессов АЦ-азного пути может усилить эффект стимула в 107 - 108 раз. Благодаря этому возможны выявление и реализация слабого сигнала, что имеет особое значение в условиях патологии, при нарушении синаптического проведения.
Многие изменения функций нейрона связаны с действием патогенных агентов на те или иные звенья указанных систем внутриклеточной сигнализации. Фармакологическая коррекция деятельности нейрона и эффекты лечебных средств также реализуются через соответствующие изменения этих систем. Так, холерный и коклюшный токсины действуют на процессы, связанные с активностью мембранных G-белков, активирующих или угнетающих АЦ-азу. Ксан-тины (теофиллин, кофеин) обусловливают накопление цАМФ, что приводит к усиленной деятельности нейрона. При действии ряда противосудо-рожных препаратов (например, дифенилгидан-тоина, карбамазепина, бензодиазепинов) и психотропных средств (например, трифтазина) угнетаются разные пути фосфорилирования бел-
ков, благодаря чему снижается активность нейронов. Ионы лития, применяемые при лечении некоторых эндогенных психозов, ослабляют деятельность системы фосфоинозитидов. С усиленным входом Са21 связана эпилептизация нейронов, блокада этого входа антагонистами Са2* подавляет эпилептическую активность.
20.3.10. Гиперактивность нейрона
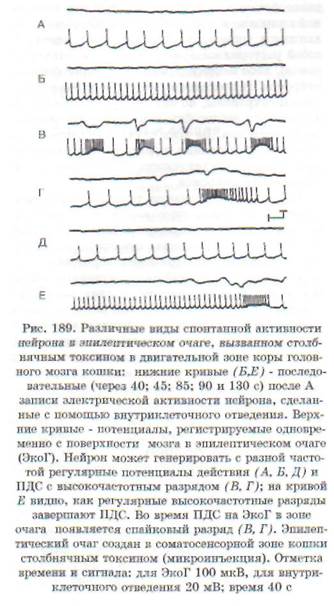
Гиперактивность нейрона обусловлена значительным, выходящим из-под контроля нарушением баланса между возбуждением и торможением нейрона в пользу возбуждения. В функциональном отношении она заключается в продуцировании нейроном усиленного потока импульсов, который может иметь различный характер: высокочастотные потенциалы действия; отдельные разряды; разряды, сгруппированные в пачки, и пр. Особый вид гиперактивности представляет собой пароксизмальный деполяризацион-ный сдвиг (ПДС) в мембране, на высоте которого возникает высокочастотный разряд (рис. 189). Такой вид гиперактивности рассматривается как проявление эпилептизации нейрона.
Указанный сдвиг баланса между возбуждением и торможением может быть обусловлен либо первичным усиленным возбуждением нейрона, преодолевающим тормозный контроль, либо первичной недостаточностью тормозного контроля. Первый механизм реализуется значительной деполяризацией мембраны и усиленным входом Na+ и Са2* в нейрон, второй - расстройством механизмов, обеспечивающих гиперполяризацию мембраны: нарушением выхода К+ из нейрона и входа С1~ в нейрон.
Существенным эндогенным регулятором активности нейрона является у-аминомасляная кислота (ГАМК). Она вызывает торможение нейрона при связывании со своим рецептором, входящим в сложный белковый дГАМК-комплекс, который состоит из нескольких субъединиц; при активации комплекса под влиянием ГАМК усиливается поступление С1~ в нейрон. При растор-маживании нейрона в связи с ослаблением гиперполяризации и деполяризацией мембраны происходит усиление поступления Са2* в нейрон. Кроме того, Са2*, находясь уже в цитозоле, нарушает поступление С1" в нейрон, ослабляя, таким образом, изнутри «ГАМКергическое» торможение. Со всеми этими путями действия Ca2f
Часть III. ПАТОФИЗИОЛОГИЯ ОРГАНОВ И СИСТЕМ
 | |||
 |
|
связана эпилептизация нейрона, возникающая под влиянием конвульсантов, которые нарушают ГАМКергическое торможение. Многие кон-вульсанты (например, пенициллин, коразол и др.) оказывают сложное действие на нейрон, одновременно активируя возбуждающие и инак-тивируя тормозные механизмы.
Хроническая стимуляция нейрона (например, при прямом электрическом раздражении, синап-гическом воздействии, под влиянием возбуждающих аминокислот и др.) даже слабой интенсивности может с течением времени привести к гиперактивации нейрона. С другой стороны,
выключение афферентации нейрона также обусловливает гиперактивацию нейрона. Этот эффект объясняется повышением чувствительности нейрона при денервации и нарушением тормозных процессов.
Таким образом, патологическая гиперактивация нейронов, их эпилептизация, представляет сложный комплекс разнообразных мембранных и внутриклеточных процессов. Для подавления эпилептической активности целесообразно комплексное применение веществ, нормализующих основные патогенетические звенья процесса. Среди корригирующих воздействий первостепенное значение имеют блокада входа Са2+ и восстановление тормозного контроля.