Главная страница Случайная страница
КАТЕГОРИИ:
АвтомобилиАстрономияБиологияГеографияДом и садДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеталлургияМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРелигияРиторикаСоциологияСпортСтроительствоТехнологияТуризмФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
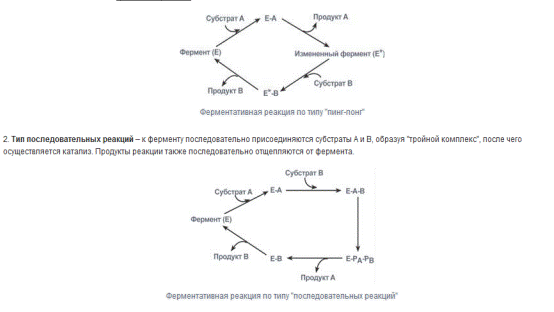
Особенности ферментативного катализа.
|
|

Ферменты.
5. После установления химической природы ферментов подтвердилось представление, выдвинутое более 80 лет назад
В. Анри, Л. Михаэлисом и М. Ментен, о том, что при энзиматическом
катализе фермент Е соединяется (в принципе обратимо) со своим субстра-
том S, образуя нестойкий промежуточный фермент-субстратный комплекс
ES, который в конце реакции распадается с освобождением фермента
и продуктов реакции Р. Благодаря высокому сродству связывания и образованию ES-комплекса резко возрастает число молекул субстрата, всту-
пающих в реакции. Эти представления легли в основу теории «ключа-
замка» Э. Фишера, которую иногда называют теорией «жесткой матрицы».
Таким образом, жесткая структура активного центра оказывается комп-
лементарной молекулярной структуре субстрата, обеспечивая тем самым
высокую специфичность фермента.
Л. Михаэлис не только постулировал образование промежуточного
фермент-субстратного ES-комплекса, но и рассчитал влияние концентрации
субстрата на скорость реакции. В процессе реакции различают несколько
стадий: присоединение молекулы субстрата к ферменту, преобразование
первичного промежуточного соединения в один или несколько последова-
тельных (переходных) комплексов и протекающее в одну или несколько
стадий отделение конечных продуктов реакции от фермента. Это можно
схематически проиллюстрировать следующими примерами:
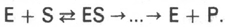
В реакциях анаболизма, например А + В —> АВ, фермент может соединяться
как с одним, так и с другим субстратом или обоими субстратами:

В реакциях катаболизма, например АВ —> А + В:
а) АВ + Е –> ABE
б) АВЕ –> А + BE
в) ВЕ –> В + Е
(а + б + в): АВ + Е –> А + В + Е
. Если фермент в активном центре содержит
кофермент, то предполагается образование тройного комплекса (рис. 4.8).
Фермент вступает во взаимодействие с субстратом на очень короткий
период, поэтому долгое время не удавалось показать образование такого
комплекса. Прямые доказательства существования фермент-субстратного
комплекса были получены в лабораториях Д. Кейлина и Б. Чанса. В на-
стоящее время экспериментальные и математические методы кинетики,
термодинамики и статической механики химических реакций позволяют
определить для ряда ферментативных реакций кинетические и термоди-
намические показатели, в частности константы диссоциации промежуточ-
ных фермент-субстратных комплексов, константы скорости и равновесия их
образования.
В образовании фермент-субстратных комплексов участвуют водородные
связи, электростатические и гидрофобные взаимодействия, а в ряде случаев
также ковалентные, координационные связи. Информация о при-
роде связей между субстратом и связывающим участком активного центра
фермента может быть получена методами ЭПР и ЯМР, а также методами
УФ- и ИК-спектроскопии.
Для каталитической активности фермента существенное значение имеет
пространственная структура, в которой жесткие участки α -спиралей че-
редуются с гибкими, эластичными линейными отрезками, обеспечиваю-
щими динамические изменения белковой молекулы фермента. Этим изме-
неням придается большое значение в некоторых теориях ферментативного
катализа. Так, в противоположность модели Э. Фишера «ключ-замок»
Д. Кошлендом была разработана теория «индуцированного соответ-
ствия», допускающая высокую конформационную лабильность молекулы
белка-фермента и гибкость и подвижность активного центра. Эта теория
была основана на весьма убедительных экспериментах, свидетельствующих
о том, что субстрат индуцирует конформационные изменения молекулы
фермента таким образом, что активный центр принимает необходимую для
связывания субстрата пространственную ориентацию. Получены экспериментальные доказательства нового
положения о том, что постулированное Д. Кошлендом «индуцированное
соответствие» субстрата и фермента создается не обязательно изменениями
конформации белковой молекулы, но также геометрической и электрон-
но-топографической перестройкой молекулы субстрата.
В каталитическом процессе существенное значение имеют точное соот-
ветствие между ферментом и субстратом, а также термодинамические
и каталитические преимущества подобного соответствия. Гипотеза «инду-
цированного соответствия» предполагает существование между ферментом
и субстратом не только пространственной или геометрической компле-
ментарности, но и электростатического соответствия, обусловленного
спариванием противоположно заряженных групп субстрата и активного
центра фермента. Точное соответствие обеспечивает образование эффек-
тивного комплекса между субстратом и ферментом.
Подобно другим катализаторам, ферменты, с термодинамической точки
зрения, ускоряют химические реакции за счет снижения энергии активации *.
Энергией активации называется энергия, необходимая для перевода
всех молекул моля вещества в активированное состояние при данной
температуре. Другими словами, это энергия, необходимая для запуска
химической реакции, без которой реакция не начинается несмотря на ее
термодинамическую вероятность. Фермент снижает энергию активации
путем увеличения числа активированных молекул, которые становятся
реакционноспособными на более низком энергетическом уровне
На рисунке видно, что ферментативная реакция имеет более низкую
энергию активации. Следует отметить, что как катализируемая ферментом,
так и не катализируемая им реакция независимо от ее пути имеет одина-
ковую величину стандартного изменения свободной энергии (Δ G). Действуя
на скорость реакции, ферменты не изменяют равновесия между прямой
и обратной реакциями, как и не влияют на величину свободной энергии
реакции; они лишь ускоряют наступление равновесия химической реакции.
Таким образом, в механизме ферментативного катализа ведущую роль
играют промежуточные фермент-субстратные комплексы, образование ко-
торых определяется как тонкой трехмерной структурой активного центра,
так и уникальной структурной организацией всей молекулы фермента,
обеспечивающими высокую каталитическую активность и специфичность
действия биокатализатора.
6. Специфичность ферментов. Ферменты обладают высокой специфич-
ностью действия. Это свойство часто существенно отличает их от неорга-
нических катализаторов. Так, мелкоизмельченные платина и палладий
могут катализировать восстановление (с участием молекулярного водо-
рода) десятков тысяч химических соединений различной структуры. Высо-
кая специфичность ферментов обусловлена, как было отмечено, конфор-
мационной и электростатической комплементарностью между молекулами
субстрата и фермента и уникальной структурной организацией активного
центра, обеспечивающими «узнавание», высокое сродство и избиратель-
ность протекания одной какой-либо реакции из тысячи других химических
реакций, осуществляющихся одновременно в живых клетках.
В зависимости от механизма действия различают ферменты с отно-
сительной (или групповой) и абсолютной специфичностью. Так,
для действия некоторых гидролитических ферментов наибольшее значение
имеет тип химической связи в молекуле субстрата. Например, пепсин
в одинаковой степени расщепляет белки животного и растительного проис-
хождения, несмотря на то что эти белки существенно отличаются друг от
друга как по химическому строению и аминокислотному составу, так и по
физико-химическим свойствам. Однако пепсин не расщепляет ни углеводы,
ни жиры. Объясняется это тем, что точкой приложения, местом действия
пепсина является пептидная —СО—NH-связь. Для действия липазы, ка-
тализирующей гидролиз жиров на глицерин и жирные кислоты, подобным
местом является сложноэфирная связь. Аналогичной групповой специфич-
ностью обладают трипсин, химотрипсин, пептидазы, ферменты, гидроли-
зующие α -гликозидные связи (но не β -гликозидные связи, имеющиеся
в целлюлозе) в полисахаридах, и др. Обычно эти ферменты участву-
ют в процессе пищеварения, и их групповая специфичность, вероятнее
всего, имеет определенный биологический смысл. Относительной специ-
фичностью наделены также некоторые внутриклеточные ферменты, на-
пример гексокиназа, катализирующая в присутствии АТФ фосфорилиро-
вание почти всех гексоз, хотя одновременно в клетках имеются и спе-
цифические для каждой гексозы ферменты, выполняющие такое же фос-
форилирование (см. главу 10).
Абсолютной специфичностью действия называют способность фермента
катализировать превращение только единственного субстрата. Любые из-
менения (модификации) в структуре субстрата делают его недоступным для
действия фермента. Примерами таких ферментов могут служить аргиназа,
расщепляющая в естественных условиях (в организме) аргинин, уреаза,
катализирующая распад мочевины, и др.
Имеются экспериментальные доказательства существования так назы-
ваемой стереохимической специфичности, обусловленной сущест-
вованием оптически изомерных L- и D-форм или геометрических (цис-
и транс-) изомеров химических веществ. Так, известны оксидазы L-
и D-аминокислот, хотя в природных белках обнаружены только L-ами-
нокислоты. Каждый из видов оксидаз действует только на свой спе-
цифический стереоизомер *.
L-аминокислота--1/2О2Оксидаза L-аминокислотà α -Кетокислота + NH3 + Н2О;
D-аминокислота---1/2О2 Оксидаза D-аминокислотà α -Кетокислота + NH3 + H2О.
Наглядным примером стереохимической специфичности является бак-
териальная аспартатдекарбоксилаза, катализирующая отщепление СО2
только от L-аспарагиновой кислоты с превращением ее в L-аланин. Сте-
реоспецифичность проявляют ферменты, катализирующие и синтетические
реакции. Так, из аммиака и α -кетоглутарата во всех живых организмах
синтезируется L-изомер глутаминовой кислоты, входящей в состав при-
родных белков. Если какое-либо соединение существует в форме цис-
и транс-изомеров с различным расположением групп атомов вокруг двой-
ной связи, то, как правило, только один из этих геометрических изомеров
может служить в качестве субстрата для действия фермента. Например,
фумараза катализирует превращение только фумаровой кислоты (транс-
изомер), но не действует на малеиновую кислоту (цис-изомер):
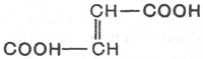
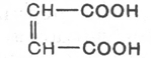
фумаровая кислота Малеиновая кислота
Таким образом, благодаря высокой специфичности действия ферменты
обеспечивают протекание с большой скоростью лишь определенных хи-
мических реакций из огромного разнообразия возможных превращений
в микропространстве клеток и целостном организме, регулируя тем самым
интенсивность обмена веществ.
7. Активность ферментов зависит от рН раствора, в котором протекает ферментативная реакция. Для каждого фермента существует значение рН, при котором наблюдается его максимальная активность. Отклонение от оптимального значения рН приводит к понижению ферментативной активности.
Влияние рН на активность ферментов связано с ионизацией функциональных групп аминокислотных остатков данного белка, обеспечивающих оптимальную конформацию активного центра фермента. При изменении рН от оптимальных значений происходит изменение ионизации функциональных групп молекулы белка. Например, при закислении среды происходит протонирование свободных аминогрупп (NH3+), а при защелачивании происходит отщепление протона от карбоксильных групп (СОО-). Это приводит к изменению конформации молекулы фермента и конформации активного центра; следовательно, нарушается присоединение субстрата, кофакторов и коферментов к активному центру. Кроме того, рН среды может влиять на степень ионизации или пространственную организацию субстрата, что также влияет на сродство субстрата к активному центру. При значительном отклонении от оптимального значения рН может происходить денатурация белковой молекулы с полной потерей ферментативной активности.
Оптимум значения рН у разных ферментов различный (рис. 2-18). Ферменты, работающие в кислых условиях среды (например, пепсин в желудке или лизосомальные ферменты), эволюционно приобретают конформацию, обеспечивающую работу фермента при кислых значениях рН. Однако большая часть ферментов организма человека имеет оптимум рН, близкий к нейтральному, совпадающий с физиологическим значением рН (табл. 2-1).
Таблица 2-1. Оптимальные значения рН для некоторых ферментов
| Фермент | Оптимальное значение рН |
| Пепсин | 1, 5-2 |
| Пируват-карбоксилаза | 4, 8 |
| Каталаза | 6, 8-7 |
| Фумараза | 6, 5 |
| Уреаза | 6, 8-7, 2 |
| Кабоксипептидаза | 7, 5 |
| Трипсин | 6, 5-7, 5 |
| Аргиназа | 9, 5-9, 9 |
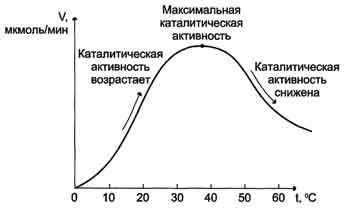
8. Повышение температуры до определённых пределов оказывает влияние на скорость ферментативной

Рис. 2-16. Зависимость скорости ферментативной реакции (V) от концентрации фермента.
реакции, подобно влиянию температуры на любую химическую реакцию. С повышением температуры ускоряется движение молекул, что приводит к повышению вероятности взаимодействия реагирующих веществ. Кроме того, температура может повышать энергию реагирующих молекул, что также приводит к ускорению реакции. Однако скорость химической реакции, катализируемая ферментами, имеет свой температурный оптимум, превышение которого сопровождается понижением ферментативной активности, возникающим из-за термической денатурации белковой молекулы (рис. 2-17).
Для большинства ферментов человека оптимальна температура 37-38 °С. Однако в природе существуют и термостабильные ферменты. Например, Taq-полимераза, выделенная из микроорганизмов, живущих в горячих источниках, не инактивируется при повышении температуры до 95 °С. Этот фермент используют в научно-практической медицине для молекулярной диагностики заболеваний с использованием метода полимеразной цепной реакции (ПЦР).
9. Зависимость скорости ферментативной реакции от количества субстрата
Если концентрацию ферментов оставить постоянной, изменяя только количество субстрата, то график скорости ферментативной реакции описывают гиперболой (рис. 2-19).
При увеличении количества субстрата начальная скорость возрастает. Когда фермент становится полностью насыщенным субстратом, т.е. происходит максимально возможное при данной концентрации фермента формирование фермент-субстратного комплекса, наблюдают наибольшую скорость образования продукта. Дальнейшее повышение концентрации субстрата не приводит к увеличению образования продукта, т.е. скорость реакции не возрастает. Данное состояние соответствует максимальной скорости реакции Vmax.
Таким образом, концентрация фермента - лимитирующий фактор в образовании продукта. Это наблюдение легло в основу ферментативной кинетики, разработанной учёными Л. Михаэлисом и М. Ментен в 1913 г.
Ферментативный процесс можно выразить следующим уравнением:

где k1 - константа скорости образования фермент-субстратного комплекса; k-1 - константа скорости обратной реакции, распада фермент-субстратного комплекса; k2 - константа скорости образования продукта реакции.
Следующее соотношение констант скоростей (k-1 + k2)/k1 называют константой Михаэлиса и обозначают Кm.
Скорость реакции пропорциональна концентрации фермент-субстратного комплекса ES, a скорость образования ES зависит от концентрации субстрата и концентрации свободного фермента. На концентрацию ES влияет скорость формирования и распада ES.
Наибольшая скорость реакции наблюдается в том случае, когда все молекулы фермента находятся в комплексе с субстратом, т.е. в фермент-субстратном комплексе ES, т.е. [Е] = [ES].
Зависимость скорости ферментативной реакции от концентрации субстрата выражается следующим уравнением (математическое выведение этой формулы можно найти в пособиях по ферментативной кинетике):
V =
| Vmax[S] |
| Km + [S] |
Это уравнение получило название уравнения Михаэлиса-Ментен.
В случае, когда скорость реакции равна половине максимальной, Km = [S] (рис. 2-19). Таким образом, константа Михаэлиса численно равна концентрации субстрата, при которой достигается половина максимальной скорости.
Уравнение Михаэлиса-Ментен - основное уравнение ферментативной кинетики, описывающее зависимость скорости ферментативной реакции от концентрации субстрата.
Если концентрация субстрата значительно больше Km (S > > Km), to увеличение концентрации субстрата на величину Кm практически не влияет на сумму (Km + S) и её можно считать равной концентрации субстрата. Следовательно, скорость реакции становится равной максимальной скорости: V = Vmax. В этих условиях реакция имеет нулевой порядок, т.е. не зависит от концентрации субстрата. Можно сделать вывод, что Vmax - величина постоянная для данной концентрации фермента, не зависящая от концентрации субстрата.
Если концентрация субстрата значительно меньше Km(S < < Km), то сумма (Km + S) примерно равна Кm, следовательно, V = Vmax[S]/Km, т.е. в данном случае скорость реакции прямо пропорциональна
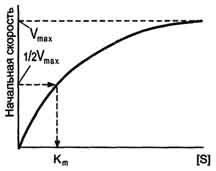
Рис. 2-19. Зависимость скорости реакции (V) от концентрации субстрата S. Vmax - максимальная скорость реакции при данной концентрации фермента в оптимальных условиях проведения реакции. Кm - константа Михаэлиса.
концентрации субстрата (реакция имеет первый порядок).
Vmах и Km - кинетические характеристики эффективности фермента.
· Vmax дает характеристику каталитической активности фермента и имеет размерность скорости ферментативной реакции моль/л, т.е. определяет максимальную возможность образования продукта при данной концентрации фермента и в условиях избытка субстрата. Кm характеризует сродство данного фермента к данному субстрату и является величиной постоянной, не зависящей от концентрации фермента. Чем меньше
· Кm, тем больше сродство фермента к данному субстрату, тем выше начальная скорость реакции и наоборот, чем больше Кm, тем меньше начальная скорость реакции, тем меньше сродство фермента к субстрату.
На рис. 2-20 представлена зависимость скорости двух ферментативных реакций (1 и 2) от концентрации субстрата. Константа Михаэлиса первого фермента меньше константы Михаэлиса второго фермента (Kml < Km2). Следовательно, сродство первого фермента к субстрату выше, чем у второго фермента, и начальная скорость реакции, катализируемой первым ферментом, выше в сравнении со вторым ферментом.
10.
11. Скорость ферментативной реакции, как и активность фермента, в значи-
тельной степени определяется также присутствием в среде активаторов
и ингибиторов: первые повышают скорость реакции, а вторые тормозят эту
реакцию. Активирующее влияние на скорость ферментативной реакции
оказывают разнообразные вещества органической и неорганической приро-
ды. Так, соляная кислота активирует действие пепсина желудочного сока;
Количество продукта
Время
Повышение
концентрации фермента
4х
3х
2х
1х
желчные кислоты повышают активность панкреатической липазы; неко-
торые тканевые ферменты (оксидоредуктазы, катепсины, аргиназа), расти-
тельная протеиназа и др. в значительной степени активируются соеди-
нениями, содержащими свободные SH-группы (глутатион, цистеин), а ряд
ферментов – также витамином С. Особенно часто активаторами выступают
ионы двухвалентных и, реже, одновалентных металлов. Получены дока-
зательства, что около четверти всех известных ферментов для проявления
полной каталитической активности нуждаются в присутствии металлов.
Многие ферменты вообще не активны в отсутствие металлов. Так, при
удалении цинка угольная ангидраза (карбоангидраза), катализирующая
биосинтез и распад Н2СО3, практически теряет свою ферментативную
активность; более того, цинк при этом не может быть заменен никаким
другим металлом. Известны ферменты *, действие которых активируется
ионами нескольких металлов; в частности, енолаза активируется Mg2+,
Mn2+, К+ (табл. 4.4).механизм действия металлов в энзиматическом катализе,
или роль металлов в активировании ферментами. В ряде случаев ионы
металлов (Со2+, Mg2+, Zn2+, Fe2+) выполняют функции простетических
групп ферментов, или служат акцепторами и донаторами электронов, или
выступают в качестве электрофилов либо нуклеофилов, сохраняя реактив-
ные группы в необходимой ориентации. В других случаях они способствуют
присоединению субстрата к активному центру и образованию фермент-
субстратного комплекса. Например, ионы Mg2+ через отрицательно заря-
женную фосфатную группу обеспечивают присоединение монофосфатных
эфиров органических веществ к активному центру фосфатаз, катализирую-
щих гидролиз этих соединений. Иногда металл соединяется с субстратом,
образуя истинный субстрат, на который действует фермент. В частности,
ионы Mg2+ активируют креатинфосфокиназу благодаря образованию ис-
тинного субстрата – магниевой соли АТФ. Наконец, имеются эксперимен-
тальные доказательства прямого участия металлов (например, ионов Са2+
* Обычно трудно провести границу между металлоферментами (когда металл связан
прочно с белком и незаменим) и ферментами, активируемыми металлами (последние лишь
ускоряют реакцию и легко диссоциируют).в молекуле амилазы слюны) в формировании и стабилизации активного
центра и всей трехмерной структуры молекулы фермента. Следует отме-
тить также, что металлы нередко выступают в роли аллостерических
модуляторов (эффекторов; см. рис. 4.22). Взаимодействуя с аллостеричес-
ким центром, подобный металл (эффектор) способствует образованию
наиболее выгодной пространственной конфигурации фермента и активного
фермент-субстратного комплекса.
Анионы в физиологических концентрациях обычно неэффективны или
оказывают небольшое активирующее влияние на ферменты. Исключение
составляют пепсин, некоторые оксидоредуктазы, активируемые анионами,
а также амилаза слюны, катализирующая гидролиз крахмала, активность
которой повышается при действии ионов хлора, и аденилатциклаза, кото-
рая активируется анионами галогенов.
12. Ингибиторы ферментов обычно принято делить на два больших
класса: обратимые и необратимые. Это вещества, вызывающие частичное
(обратимое) или полное торможение реакций, катализируемых фермента-
ми. Недавно открыты антиферменты (антиэнзимы, или антизимы),
представляющие собой белки (или полипептиды), действующие как инги-
биторы ферментов. К подобным веществам относятся, например, инги-
битор трипсина, обнаруженный в соевых бобах, и сывороточный анти-
трипсин. Недавно открыт в печени животных антифермент орнитинде-
карбоксилазы (см. главу 12). Антизимы, вероятнее всего, образуют трудно-
диссоциируемые комплексы с соответствующими ферментами, выключая
их из химических реакций. Иногда ингибитор является составным компо-
нентом предшественника фермента, например пепсина (см. главу 12), или
входит в состав сложных комплексов ферментов, например в состав
протеинкиназы и протеинфосфатазы, катализирующих процессы фосфо-
рилирования-дефосфорилирования в живых организмах. Однако до сих
пор не выяснено, являются ли подобные антиферменты истинными инги-
биторами или регуляторными субъединицами, в частности, какова разница
в назначении регуляторной (R) субъединицы в составе протеинкиназы
и ингибиторной (I) субъединицы в составе протеинфосфатазы.
Ферменты являются белками, поэтому любые агенты, вызывающие
денатурацию белка (кислоты, щелочи, соли тяжелых металлов, нагревание),
приводят к необратимой инактивации фермента. Однако подобное инак-
тивирование относительно неспецифично, оно не связано с механизмом
действия ферментов. Гораздо большую группу составляют так называемые
специфические ингибиторы, которые оказывают свое действие на какой-
либо один фермент или группу родственных ферментов, вызывая обрати-
мое или необратимое ингибирование. Исследование этих ингибиторов
имеет важное значение. Во-первых, ингибиторы могут дать ценную инфор-
мацию о химической природе активного центра фермента, а также о составе
его функциональных групп и природе химических связей, обеспечивающих
образование фермент-субстратного комплекса. Известны вещества, вклю-
чая лекарственные препараты, специфически связывающие ту или иную
функциональную группу в молекуле фермента, выключая ее из химической
реакции. Так, йодацетат IСН2—СООН, его амид и этиловый эфир, пара-
хлормеркурибензоат ClHg—С6Н4—СООН и другие реагенты сравнитель-
но легко вступают в химическую связь с некоторыми SH-группами фер-
ментов. Если такие группы имеют существенное значение для акта ката-
лиза, то добавление подобных ингибиторов приводит к полной потере
активности фермента:
R-SH + IСН2—СООН —> НI + R—S—CH2—COOH
Действие ряда других ферментов (холинэстераза, трипсин и химотрип-
син) сильно тормозится некоторыми фосфорорганическими соединениями,
например ДФФ, вследствие блокирования ключевой гидроксильной группы
серина в активном центре (см. ранее).
Во-вторых, ингибиторы нашли широкое применение в энзимологии при
исследовании природы множественных форм ферментов и изоферментов,
различающихся не столько электрофоретической подвижностью, сколько
различной чувствительностью к одному и тому же ингибитору.
При помощи ингибиторов, выключающих отдельные стадии многосту-
пенчатого метаболического процесса, могут быть точно установлены не
только последовательность химических реакций, но и природа участвую-
щих в этих превращениях ферментов. Этим путем, применяя йодацетат,
фториды и другие специфические ингибиторы, был расшифрован глико-
литический путь окислительно-восстановительных превращений глюкозы
до стадии образования молочной кислоты в мышечной ткани, насчиты-
вающий 11 стадий с участием 11 ферментов и 10 промежуточных ме-
таболитов.
С ингибированием ферментов связан механизм действия многих токси-
нов и ядов на организм. Известно, что при отравлениях солями сенильной
кислоты смерть наступает вследствие полного торможения и выключения
дыхательных ферментов (цитохромная система) тканей, особенно клеток
мозга. Токсическое влияние на организм человека и животных некоторых
инсектицидов обусловлено торможением активности холинэстеразы – фер-
мента, играющего ключевую роль в деятельности нервной системы.
Современная, так называемая рациональная, химиотерапия (направлен-
ное применение лекарственных препаратов в медицине) должна основы-
ваться на точном знании механизма действия лекарственных средств на
биосинтез ферментов, на активность уже синтезированных ферментов или
на регуляцию их активности в организме. Иногда для лечения некоторых
болезней используют избирательно действующие ингибиторы. Так, инги-
битор ряда протеиназ (трипсина, химотрипсина и калликреина) трасилол
широко применяется для лечения острого панкреатита – болезни, при ко-
торой уровень трипсина и химотрипсина в крови резко возрастает. Знание
избирательного ингибиторного действия некоторых природных и синте-
тических соединений (так называемых антиметаболитов) на ферменты
может служить методологической основой для разработки эффективных
методов синтеза химиотерапевтических препаратов. Этот путь открывает
широкие возможности для направленного воздействия на синтез ферментов
в организме и регуляции интенсивности метаболизма при патологии.
Типы ингибирования. Различают обратимое и необратимое ингибиро-
вание. Если ингибитор вызывает стойкие изменения пространственной
третичной структуры молекулы фермента или модификацию функциональ-
ных групп фермента, то такой тип ингибирования называется необрати-
мым. Чаще, однако, имеет место обратимое ингибирование, под-
дающееся количественному изучению на основе уравнения Михаэлиса-
Ментен. Обратимое ингибирование в свою очередь разделяют на кон-
курентное и неконкурентное в зависимости от того, удается или не удается
преодолеть торможение ферментативной реакции путем увеличения кон-
центрации субстрата.
Конкурентное ингибирование.
Неконкурентное ингибирование вызывается веществами, не
имеющими структурного сходства с субстратами и часто связывающимися
не с активным центром, а в другом месте молекулы фермента. Степень
торможения во многих случаях определяется продолжительностью дейст-
вия ингибитора на фермент. При данном типе ингибирования благодаря
образованию стабильной ковалентной связи фермент часто подвергается
полной инактивации, и тогда торможение становится необратимым. При-
мером необратимого ингибирования является действие йодацетата, ДФФ,
а также диэтил-n-нитрофенилфосфата и солей синильной кислоты. Это
действие заключается в связывании и выключении функциональных групп
или ионов металлов и молекуле фермента.
Следует указать, что неконкурентное ингибирование также может быть
обратимым и необратимым, поскольку отсутствует конкуренция между
субстратом и ингибитором за активный центр. Примеры необратимого
ингибирования приведены ранее. При обратимом неконкурентном
ингибировании субстрат S и ингибитор I связываются с разными
центрами, поэтому появляется возможность образования как комплекса EI,
так и тройного комплекса EIS; последний может распадаться с осво-
бождением продукта, но с меньшей скоростью, чем комплекс ES.
Этот тип неконкурентного ингибирования чаще всего наблюдается
у ферментов, катализирующих превращения более одного субстрата, когда
связывание ингибитора не блокирует связывание субстрата с активным
центром. Ингибитор при этом соединяется как со свободным ферментом,
так и с ES-комплексом.
Известно, кроме того, так называемое бесконкурентное ингиби-
рование, когда ингибитор связывается с ферментом также в некатали-
тическом центре, однако не со свободным ферментом, а только с ES-комп-
лексом в виде тройного комплекса.
Для выяснения вопроса о типе ингибирования пользуются уравнениями
Михаэлиса-Ментен, Лайнуивера-Бэрка или другими, например уравне-
нием Эди-Хофсти:
v = –Km(v/[S]) + Vmax
и соответствующими графиками в прямолинейных координатах.
При конкурентном типе ингибирования ингибитор увеличивает значение
Кm, не оказывая влияния на максимальную скорость Vmax (рис. 4.21). Это
означает, что при достаточно высокой концентрации субстрата [S] ин-
гибитор вытесняется молекулами субстрата из комплекса EI. При некон-
курентном ингибировании (рис. 4.22) ингибитор снижает величину макси-
мальной скорости. Если при этом величина Кm не уменьшается, то говорят
о полностью неконкурентном ингибировании. Подобный тип ингибиро-
вания имеет место при образовании неактивных, труднодиссоциирующих
комплексов EI и(или) EIS. Часто, однако, наблюдается смешанный тип
ингибирования, иногда называемый частично неконкурентным, или обра-
тимым неконкурентным ингибированием (см. ранее), при котором сни-
жение Vmax сочетается с одновременным увеличением значений Кm. Это
означает, что комплекс EI сохраняет частичную активность, т.е. способ-
ность к образованию промежуточного тройного комплекса EIS, в котором
субстрат подвергается
13.
Конкурентное ингибирование может быть вызвано веществами,
имеющими структуру, похожую на структуру субстрата, но несколько
отличающуюся от структуры истинного субстрата. Такое ингибирование
основано на связывании ингибитора с субстратсвязывающим (активным)
центром. Классическим примером подобного типа ингибирования является
торможение сукцинатдегидрогеназы (СДГ) малоновой кислотой. Этот фер-
мент катализирует окисление путем дегидрирования янтарной кислоты
(сукцината) в фумаровую:
Если в среду добавить малонат (ингибитор), то в результате структур-
ного сходства его с истинным субстратом сукцинатом (наличие двух таких
же ионизированных карбоксильных групп) он будет взаимодействовать
с активным центром с образованием фермент-ингибиторного комплекса,
однако при этом полностью исключается перенос атома водорода от
малоната. Структуры субстрата (сукцинат) и ингибитора (малонат) все же
несколько различаются. Поэтому они конкурируют за связывание с актив-
ным центром, и степень торможения будет определяться соотношением
концентраций малоната и сукцината, а не абсолютной концентрацией
ингибитора. Таким образом, ингибитор может обратимо связываться
с ферментом, образуя фермент-ингибиторный комплекс. Этот тип ингиби-
рования иногда называют ингибированием по типу метаболического анта-
гонизма.
В общей форме реакция взаимодействия ингибитора с ферментом может
быть представлена следующим уравнением:
Образовавшийся комплекс, называемый фермент-ингибиторным комп-
лексом ЕI, в отличие от фермент-субстратного комплекса ES не распадается
с образованием продуктов реакции. Константу диссоциации комплекса EI,
или ингибиторную константу Кi, можно, следуя теории Михаэлиса–Мен-
тен, определить как отношение констант обратной и прямой реакций:
т.е. ингибиторная константа прямо пропорциональна произведению кон-
центрации фермента и ингибитора и обратно пропорциональна концент-
рации комплекса EI.
Метод конкурентного торможения нашел широкое применение в ме-
дицинской практике. Известно, например, что для лечения некоторых
инфекционных заболеваний, вызываемых бактериями, применяют сульфа-
ниламидные препараты. Оказалось, что эти препараты имеют структурное
сходство с парааминобензойной кислотой, которую бактериальная клетка
использует для синтеза фолиевой кислоты, являющейся составной частью
ферментов бактерий. Благодаря этому структурному сходству сульфани-
ламид блокирует действие фермента путем вытеснения парааминобензой-
ной кислоты из комплекса с ферментом, синтезирующим фолиевую кисло-
ту, что ведет к торможению роста бактерий.
Некоторые аналоги витамина В6 и фолиевой кислоты, в частности
дезоксипиридоксин и аминоптерин (см. главу 7), действуют как конкурент-
ные, так называемые коферментные, ингибиторы (или антивитамины),
тормозящие многие интенсивно протекающие при патологии биологи-
ческие процессы в организме. Применение подобных аналогов в меди-
цинской практике (в частности, в дерматологии и онкологии) основано на
конкурентном вытеснении коферментов из субстратсвязывающих центров
ключевых ферментов обмена.
14. неконкурентное
Неконкурентное ингибирование вызывается веществами, не
имеющими структурного сходства с субстратами и часто связывающимися
не с активным центром, а в другом месте молекулы фермента. Степень
торможения во многих случаях определяется продолжительностью дейст-
вия ингибитора на фермент. При данном типе ингибирования благодаря
образованию стабильной ковалентной связи фермент часто подвергается
полной инактивации, и тогда торможение становится необратимым. При-
мером необратимого ингибирования является действие йодацетата, ДФФ,
а также диэтил-n-нитрофенилфосфата и солей синильной кислоты. Это
действие заключается в связывании и выключении функциональных групп
или ионов металлов и молекуле фермента.
Следует указать, что неконкурентное ингибирование также может быть
обратимым и необратимым, поскольку отсутствует конкуренция между
субстратом и ингибитором за активный центр. Примеры необратимого
ингибирования приведены ранее. При обратимом неконкурентном
ингибировании субстрат S и ингибитор I связываются с разными
центрами, поэтому появляется возможность образования как комплекса EI,
так и тройного комплекса EIS; последний может распадаться с осво-
бождением продукта, но с меньшей скоростью, чем комплекс ES.
Е + S à E Sà E+P
+ +
I I
Е I + Sà E SI
Этот тип неконкурентного ингибирования чаще всего наблюдается
у ферментов, катализирующих превращения более одного субстрата, когда
связывание ингибитора не блокирует связывание субстрата с активным
центром. Ингибитор при этом соединяется как со свободным ферментом, так и с ES комплексом.
15. Ингибиторы-лекарства,
Лекарственные препараты, применяемые с целью подавления активности ферментов, называются ингибиторами ферментов.
Классификация
1. Ингибиторы протеиназ: контрикал.
2. Ингибиторы фибринолиза: кислота амино-капроновая.
3. Антихолинэстеразные средства: прозерин, физостигмина салицилат, галантамина гидробромид и др.
4. Ингибиторы МАО: ниаламид.
5. Ингибиторы карбоангидразы: диакарб.
6. Ингибиторы ксантиноксидазы: аллопуринол.
7. Ингибиторы ацетальдегидрогеназы: циамид, тетурам (дисульфирам) и др.
Контрикал — антиферментный препарат, ингибирующий активность трипсина, калликреина, плазмина.
Фармакокинетика: при внутривенном введении действие развивается через 10—15 мин.
Показания к применению: острый панкреатит, панкреанекроз в сочетании с гепарином в острый период инфаркта миокарда.
Противопоказания: с осторожностью у лиц, склонных к аллергическим реакциям.
Побочные эффекты: аллергические реакции.
16. Под коферментом часто подразумевают дополнительную группу,
легко отделяемую от апофермента при диссоциации. Предполагают, что
простетическая группа может быть связана с белком ковалентными и неко-
валентными связями. Так, в молекуле ацетилкоэнзим-А-карбоксилазы ко-
фактор биотин ковалентно связан с апоферментом посредством амидной
связи (см. главу 7). С другой стороны, химические связи между кофакто-
рами и пептидными цепями могут быть относительно слабыми (например,
водородные связи, электростатические взаимодействия и др.). В таких
случаях при выделении ферментов наблюдается полная диссоциация обеих
частей, и изолированый белковый компонент оказывается лишенным фер-
ментативной активности, пока не будет добавлен извне недостающий
кофактор. Именно к подобным изолированным низкомолекулярным орга-
ническим веществам применим термин «кофермент», типичными предста-
вителями которых являются витамины В1, В2, В6, РР, содержащие кофер-
менты. Известно также, что и простетические группы, и коферменты
активно включаются в химические реакции, выполняя функции промежу-
тоных переносчиков электронов, атомов водорода или различных функцио-
нальных групп (например, аминных, ацетильных, карбоксильных). В подоб-
ных случаях кофермент рассматривают в качестве второго субстрата, или
косубстрата.
Роль кофермента (Ко) в качестве переносчика, например, атомов водо-
рода может быть представлена в виде схемы, где SH – субстрат, КоЕ – хо-
лофермент, А – акцептор протона:
S H КoЕ A H
S КоЕН A
Субстрат подвергается окислению, отдавая электроны и протоны,
а КоЕ – восстановлению, принимая электроны и протоны. В следующей
полуреакции восстановленный КоЕН может отдавать электроны и протоны
на какой-либо другой промежуточный переносчик электронов и протонов
или на конечный акцептор (см. главу 9).
Коэнзим, кофактор, простетическая группа – двусмысленный биохими-
ческий жаргон. До сих пор продолжается терминологический спор, по-
скольку часто определения «коэнзим», «кофактор» и «простетическая груп-
па» рассматриваются через призму их роли в реакциях энзиматического
(ферментативного) катализа. Следует, однако, считаться с тем неоспо-
римым фактом, что во многих случаях небелковые органические молекулы,
как и ионы металлов, абсолютно необходимы белковому компоненту при
выполнении определенной биологической функции, не имеющей отношения
к биокатализу. Несомненно, имеют значение также тип и характер связи
небелкового компонента с молекулой белка. Поэтому очевидно, что ко-
фактором может служить любой фактор, абсолютно необходимый для
выполнения белком его каталитической или любой другой биологической
роли. С другой стороны, коферментом может быть любой небелковый
фактор, который непосредственно вовлечен в реакцию энзиматического
катализа. Кофактор, который непосредственно не участвует в акте ката-
лиза, не является коэнзимом. В то же время простетическую группу
(ковалентно связанный небелковый компонент, необходимый для опреде-
ленной функции) можно назвать коферментом, если она непосредственно
участвует в энзиматической реакции. Простетическая группа, которая не
вовлечена в акт катализа, но функционально является существенным как
для фермента, так и для некаталитического белка, может быть названа
кофактором. И наконец, кофактор и кофермент, непрочно связанные (или
слабо связанные) с ферментом или белком, тем не менее не классифи-
цируются в качестве простетических групп.
Многие двухвалентные металлы (Mg2+, Мn2+, Са2+), как будет пока-
зано далее, также выполняют роль кофакторов, хотя они не относятся ни
к коферментам, ни к простетическим группам. Известны примеры, когда
ионы металлов прочно связаны с белковой молекулой, выполняя функции
простетической группы. В частности, очищенный фермент, катализирую-
щий окисление аскорбиновой кислоты (витамин С) в дезоксиаскорбиновую
кислоту, содержит 8 атомов меди на одну молекулу; все они настолько
прочно связаны с белковой молекулой, что даже не обмениваются с ионо-
обменными смолами и не отделяются методом диализа. Более того,
с помощью метода электронного парамагнитного резонанса показано
участие ионов меди в промежуточном переносе электронов. Интересно
отметить, что свободные ионы меди также наделены каталитической
активностью при окислении аскорбиновой кислоты, однако эта активность
повышается во многие тысячи раз, если ионы меди соединяются с апофер-
ментом в единый комплекс – холофермент.
Данные о важнейших коферментах и простетических группах ферментов,
включая их наименования и структуру, химическую природу витамина,
входящего в их состав, и характер выполняемой биохимической функции
в метаболизме, детально рассмотрены в главах 7 и 9–13.
Получены доказательства кофакторной функции в ферментативных
реакциях и ряда других биологически активных соединений, не относящихся
к витаминам: HS-глутатиона, АТФ, липоевой кислоты, производных ну-
клеозидов (уридинфосфат, цитидинфосфат, фосфоаденозинфосфосульфат),
порфиринсодержащих веществ и др. Сюда же могут быть отнесены тРНК,
которые в составе ферментов аминоацил-тРНК-синтетаз принимают ак-
тивное участие в транспорте аминокислот в рибосоме, где осуществляется
синтез белка (см. главу 14).
Следует отметить одну отличительную особенность двухкомпонентных
ферментов: ни кофактор отдельно (включая большинство коферментов), ни
сам по себе апофермент каталитической активностью не наделены, и только
их объединение в одно целое, протекающее не хаотично, а в соответствии
с программой их структурной организации, обеспечивает быстрое про-
текание химической реакции.
17. Коферменты:
По химической природе: витаминные, витаминоподобные, невитаминные.
По механизму действия: переносчики атомов водорода, электронов, протонов.
переносчики отдельных химических групп.
18. Над и НАДФ
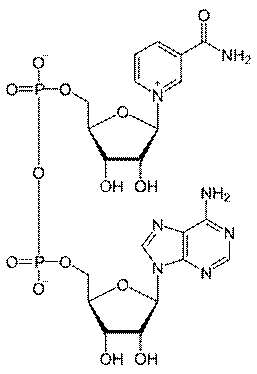
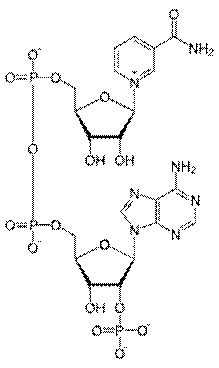
Производные PP витамина. Биохимическая функция: дыхание, перенос водорода.
Витамин РР входит в состав НАД или НАДФ,
являющихся коферментами большого числа обратимо действующих в
окислительно-восстановительных реакциях дегидрогеназ (формулы ко-
ферментов приведены в главе 9).
Показано, что ряд дегидрогеназ использует только НАД и НАДФ
(соответственно малатдегидрогеназа и глюкозо-6-фосфатдегидрогеназа),
другие могут катализировать окислительно-восстановительные реакции
в присутствии любого из них (например, глутаматдегидрогеназа; см. главу
12). В процессе биологического окисления НАД и НАДФ выполняют роль
промежуточных переносчиков электронов и протонов между окисляемым
субстратом и флавиновыми ферментами (молекулярные механизмы учас-
тия пиридиновых нуклеотидов в этом процессе подробно рассматриваются
19. ФАД и ФМН


Производные витамина B2.Биологическая функция дыхание, перенос водорода.
Рибофлавин входит в состав флавиновых кофер-
ментов, в частности ФМН и ФАД *, являющихся в свою очередь просте-
тическими группами ферментов ряда других сложных белков – флаво-
протеинов. Некоторые флавопротеины в дополнение к ФМН или ФАД
содержат еще прочно связанные неорганические ионы, в частности железо
или молибден, наделенные способностью катализировать транспорт элек-
тронов. Различают 2 типа химических реакций, катализируемых этими
ферментами. К первому относятся реакции, в которых фермент осуществ-
ляет прямое окисление с участием кислорода, т.е. дегидрирование (от-
щепление электронов и протонов) исходного субстрата или промежуточ-
ного метаболита. К ферментам этой группы относятся оксидазы L- и
D-аминокислот, глициноксидаза, альдегидоксидаза, ксантиноксидаза и др.
Вторая группа реакций, катализируемых флавопротеинами, характеризует-
ся переносом электронов и протонов не от исходного субстрата, а от
восстановленных пиридиновых коферментов. Ферменты этой группы иг-
рают главную роль в биологическом окислении. В каталитическом цикле
изоаллоксазиновый остаток ФАД или ФМН подвергается обратимому
восстановлению с присоединением электронов и атомов водорода к N1
и N10. ФМН и ФАД прочно связываются с белковым компонентом, иногда
даже ковалентно, как, например, в молекуле сукцинатдегидрогеназы.
ФМН синтезируется в организме животных из свободного рибофлавина
и АТФ при участии специфического фермента рибофлавинкиназы:
Образование ФАД в тканях также протекает при участии специфического
АТФ-зависимого фермента ФМН-аденилилтрансферазы. Исходным ве-
ществом для синтеза является ФМН:
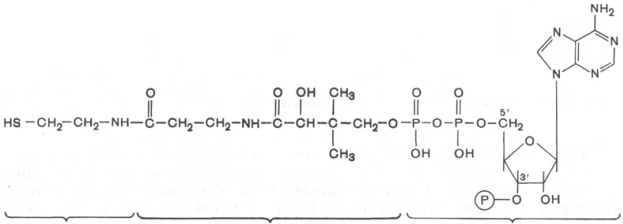
20. КоА
Витамин В3, транспорт ацильных групп.
Пантотеновая кислота входит в состав кофер-
мента А, или коэнзима А (КоА). Название «коэнзим А» (кофермент
ацилирования) связано с тем, что это соединение участвует в фермен-
тативных реакциях, катализирующих как активирование, так и перенос
ацетильного радикала СН3СО; позже оказалось, что КоА активирует
и переносит также другие кислотные остатки (ацилы). В результате обра-
зования ацил-КоА происходит активация карбоновой кислоты, которая
поднимается на более высокий энергетический уровень, создающий вы-
годные термодинамические предпосылки для ее использования в реакциях,
протекающих с потреблением энергии.
Строение КоА расшифровал Ф. Линен. В основе структуры лежит
остаток 3'-фосфоаденозин-5'-дифосфата (отличается от АТФ наличием у
3'-гидроксила фосфатной группы), соединенный с остатком пантотеновой
кислоты, карбонильная группа которой в свою очередь связана с остатком
β -меркаптоэтиламина (тиоэтиламина).
Реакционноспособным участком молекулы КоА в биохимических реак-
циях является SH-группа, поэтому принято сокращенное обозначение КоА
в виде SH-KoA. О важнейшем значении КоА в обмене веществ (как будет
показано далее – см. главы 9–11) свидетельствуют обязательное непосред-
ственное участие его в основных биохимических процессах, окисление
и биосинтез высших жирных кислот, окислительное декарбоксилирование
α -кетокислот (пируват, α -кетоглутарат), биосинтез нейтральных жиров,
фосфолипидов, стероидных гормонов, гема гемоглобина, ацетилхолина,
гиппуровой кислоты и др.
21. Тиаминпирофосфат
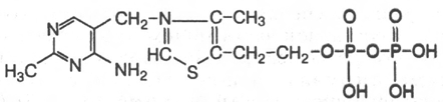
В1, декарбоксилирование а-кетокислот, перенос активного альдегида (транскетолаза).
Экспериментально доказано, что витамин B1 в
форме ТПФ является составной часть минимум 5 ферментов, участвующих
в промежуточном обмене веществ. ТПФ входит в состав двух сложных
ферментных систем – пируват- и α -кетоглутаратдегидрогеназных
комплексов, катализирующих окислительное декарбоксилирование
пировиноградной и α -кетоглутаровой кислот. В составе транскетолазы
ТПФ участвует в переносе гликоальдегидного радикала от кетосахаров на
альдосахара (см. главу 10). ТПФ является коферментом пируватдекар-
боксилазы клеток дрожжей (при алкогольной ферментации) и дегидро-
геназы γ -оксикетоглутаровой кислоты.
Приведенными примерами, вероятнее всего, не ограничиваются биоло-
гические функции тиамина. В частности, ТПФ участвует в окислительном
декарбоксилировании глиоксиловой кислоты и α -кетокислот, образующих-
ся при распаде аминокислот с разветвленной боковой цепью; в растениях
ТПФ является эссенциальным кофактором при синтезе валина и лейцина
в составе фермента ацетолактатсинтетазы.
22. Пиродаксальфосфат.
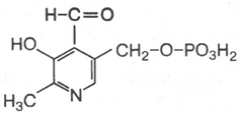
В6, обмена аминокислот, перенос аминогрупп.
Оказалось, что, хотя все три производных 3-окси-
пиридина наделены витаминными свойствами, коферментные функции
выполняют только фосфорилированные производные пиридоксаля и пи-
ридоксамина.
Фосфорилирование пиридоксаля и пиридоксамина является фермен-
тативной реакцией, протекающей при участии специфических киназ. Синтез
пиридоксальфосфата, например, катализирует пиридоксалькиназа, которая
наиболее активна в ткани мозга. Эту реакцию можно представить сле-
дующим уравнением:
Пиридоксаль + АТФ –> Пиридоксальфосфат + АДФ.
Доказано, что в животных тканях происходят взаимопревращения
пиридоксальфосфата и пиридоксаминфосфата, в частности в реакциях
трансаминирования и декарбоксилирования аминокислот (см. главу 12).
Следует отметить, что в выяснение биологической роли витамина В6
и пиридоксальфосфата в азотистом обмене существенный вклад внесли
А.Е. Браунштейн, С.Р. Мардашев, Э. Снелл, Д. Мецлер, А. Майстер и др.
Известно более 20 пиридоксалевых ферментов, катализирующих ключевые
реакции азотистого метаболизма во всех живых организмах. Так доказано,
что пиридоксальфосфат является простетической группой аминотранс-
фераз, катализирующих обратимый перенос аминогруппы (NH2-группы)
от аминокислот на α -кетокислоту, и декарбоксилаз аминокислот, осу-
ществляющих необратимое отщепление СО2 от карбоксильной группы
аминокислот с образованием биогенных аминов. Установлена кофер-
ментная роль пиридоксальфосфата в ферментативных реакциях неокисли-
тельного дезаминирования серина и треонина, окисления триптофана,
кинуренина, превращения серосодержащих аминокислот, взаимопревраще-
ния серина и глицина (см. главу 12), а также в синтезе δ -аминолевулиновой
кислоты, являющейся предшественником молекулы гема гемоглобина, и др.
Пиридоксин относится к витаминам, коферментная роль которых изучена
наиболее подробно. В последние годы число вновь открытых пиридокса-
левых ферментов быстро увеличивалось. Так, для действия гликогенфос-
форилазы существенной оказалась фосфорильная, а не альдегидная группа
пиридоксальфосфата. Вследствие широкого участия пиридоксальфосфата
в процессах обмена при недостаточности витамина В6 отмечаются разно-
образные нарушения метаболизма аминокислот.
23. Тетрагидрофолиевая кислота.
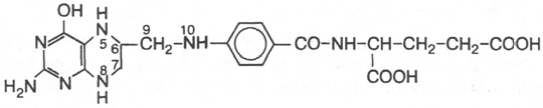
Фолиевая кислота, Транспорт одноуглеродных групп.
Коферментные функции фолиевой кислоты связаны
не со свободной формой витамина, а с восстановленным его птеридиновым
производным. Восстановление сводится к разрыву двух двойных связей
и присоединению четырех водородных атомов в положениях 5, 6, 7 и 8
с образованием тетрагидрофолиевой кислоты (ТГФК). Оно протекает
в 2 стадии в животных тканях при участии специфических ферментов,
содержащих восстановленный НАДФ. Сначала при действии фолатредук-
тазы образуется дигидрофолиевая кислота (ДГФК), которая при участии
второго фермента – дигидрофолатредуктазы – восстанавливается в ТГФК:
ФК + НАДФН + Н+ < => ДГФК + НАДФ+;
ДГФК + НАДФН + Н+ < => ТГФК + НАДФ+
Доказано, что коферментные функции ТГФК непосредственно связаны
с переносом одноуглеродных групп, первичными источниками которых
в организме являются β -углеродный атом серина, α -углеродный атом
Фолиевая (птероилглутаминовая) кислота
5, 6, 7, 8-Тетрагидрофолиевая кислота (ТГФК)
глицина, углерод метальных групп метионина, холина, 2-й углеродный
атом индольного кольца триптофана, 2-й углеродный атом имидазольного
кольца гистидина, а также формальдегид, муравьиная кислота и метанол.
К настоящему времени открыто шесть одноуглеродных групп, включа-
ющихся в разнообразные биохимические превращения в составе ТГФК:
формильная (—СНО), метильная (—СН3), метиленовая (—СН2—),
метенильная (—СН=), оксиметильная (—СН2ОН) и формими-
новая (—CH=NH). Выяснено, что присоединение этих фрагментов к
ТГФК является ферментативной реакцией ковалентного связывания их
с 5-м или 10-м атомом азота (или с обоими атомами вместе). В качестве
примера приводим отдельные функциональные группы в активных участках
ТГФК:
Имеются данные, что производные ТГФК участвуют в переносе одно-
углеродных фрагментов при биосинтезе метионина и тимина (перенос
метильной группы), серина (перенос оксиметильной группы), образовании
пуриновых нуклеотидов (перенос формильной группы) и т.д. (см. главы 12
и 13). Перечисленные вещества играют исключительно важную, ключевую,
роль в биосинтезе белков и нуклеиновых кислот, поэтому становятся
понятными те глубокие нарушения обмена, которые наблюдаются при
недостаточности фолиевой кислоты.
В медицинской практике (в частности, в онкологии) нашли применение
некоторые синтетические аналоги (антагонисты) фолиевой кислоты. Так,
4-аминоптерин используется в качестве препарата, тормозящего синтез
нуклеиновых кислот, и рекомендуется в качестве лечебного препарата при
опухолевых поражениях, в частности при острых и хронических формах
лейкозов у детей и взрослых.
24. Биотин.
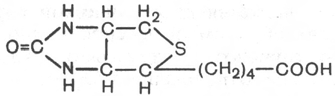
Витамин Н, кофермент карбоксилирования.
Биотин подробно изучен благодаря работам
Ф. Линена. Известные к настоящему времени биотиновые ферменты (т.е.
ферменты, содержащие в качестве кофермента биотин) катализируют два
типа реакций:
1) реакции карбоксилирования (с участием СО2 или НСО3
–), сопря-
женные с распадом АТФ
RH + HCO3
– + АТФ < => R-COOH + АДФ + Н3РО4;
2) реакции транскарбоксилирования (протекающие без участия АТФ),
при которых субстраты обмениваются карбоксильной группой
R1-COOH + R2H < => R1H + R2-COOH.
Получены доказательства двустадийного механизма этих реакций с
образованием промежуточного комплекса (карбоксибиотинилфермент).
К реакциям первого типа относятся, например, ацетил-КоА- и пируват-
карбоксилазные реакции:
CH3–CO–S-KoA + CO2 + АТФ < => HOOC–CH2–CO–KoA + АДФ + Pi.
Пируваткарбоксилаза является высокоспецифичным ферментом, ката-
лизирующим уникальную реакцию усвоения СО2 в организме животных.
Сущность реакции сводится к пополнению запасов оксалоацетата (щаве-
левоуксусная кислота) в лимоннокислом цикле (так называемые «анаплеро-
тические», «пополняющие» реакции), т.е. его синтезу из СО2 и пирувата:
Пируват + CO2 + АТФ + H2O —> Оксалоацетат + АДФ + Pi + 2H+
Реакция протекает в две стадии: на первой стадии, связанной с затратой
энергии, СО2 подвергается активированию, т.е. ковалентному связыванию
с биотином в активном центре фермента (Е-биотин):
CO2 + Биотин + АТФ + H2O Биотин + АДФ + Pi + 2H+
На второй стадии СО2 из комплекса переносится на пируват с об-
разованием оксалоацетата и освобождением фермента:
Примером второго типа реакций является метилмалонил-оксалоаце-
тат-транскарбоксилазная реакция, катализирующая обратимое превраще-
ние пировиноградной и щавелевоуксусной кислот:
Реакции карбоксилирования и транскарбоксилирования имеют важное
значение в организме при синтезе высших жирных кислот, белков, пури-
новых нуклеотидов (соответственно нуклеиновых кислот) и др.
25. Активный центр ферментов
При изучении механизма химической реакции, катализируемой фермента-
ми, исследователя всегда интересует не только определение промежуточных
и конечных продуктов и выяснение отдельных стадий реакции, но и природа
тех функциональных групп в молекуле фермента, которые обеспечивают
специфичность действия фермента на данный субстрат (субстраты) и высо-
кую каталитическую активность. Речь идет, следовательно, о точном
знании геометрии и третичной структуры фермента, а также химической
природы того участка (участков) молекулы фермента, который обеспе-
чивает высокую скорость каталитической реакции. Участвующие в фер-
ментативных реакциях молекулы субстратов часто имеют небольшие раз-
меры по сравнению с молекулами ферментов, поэтому было высказано
предположение, что при образовании фермент-субстратных комплексов
в непосредственный контакт с молекулой субстрата, очевидно, вступает
ограниченная часть аминокислот пептидной цепи. Отсюда возникло пред-
ставление об активном центре фермента. Под активным центром
подразумевают уникальную комбинацию аминокислотных остатков в мо-
лекуле фермента, обеспечивающую непосредственное связывание ее с мо-
лекулой субстрата и прямое участие в акте катализа (рис. 4.2). Установлено,
что у сложных ферментов в состав активного центра входят также просте-
тические группы.
В активном центре условно различают так называемый каталити-