Главная страница Случайная страница
КАТЕГОРИИ:
АвтомобилиАстрономияБиологияГеографияДом и садДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеталлургияМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРелигияРиторикаСоциологияСпортСтроительствоТехнологияТуризмФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Вивчення асортименту та оцінка якості рослинних олій
|
|
ДОСЛІДЖЕННЯ ХАРЧОВИХ ЖИРІВ
Завдання 1. Вивчити асортимент рослинних олій.
Для виконання завдання слід вивчити ГОСТ 1129-93 «Масло подсолнечное. Технические условия»; ГОСТ 8808-91 «Масло кукурузное. Технические условия»; ГОСТ 1128-75 «Масло хлопковое рафинированное. Технические условия»; ГОСТ 7825-96 «Масло соевое. Технические условия»; ГОСТ 8988-77 «Масло рапсовое. Технические условия»; ГОСТ 7981-68 «Масло арахисовое. Технические условия»; ГОСТ 8807-94 «Масло горчичное»; ГОСТ 10766-64 «Масло кокосовое. Технические условия»; ГОСТ 5791-81 «Масло льняное»; ГОСТ 8990-59 «Масло кунжутное (сезамовое)»; ГОСТ 8989-73 «Масло конопляное».
Завдання 2. Вивчити правила відбору проб.
Відбір проб провадять за ГОСТ 5471-83 «Масла растительные. Правила приемки и методы отбора проб». При цьому від кожної однорідної партії олії відбирають середню пробу. Середньою пробою вважається частина олії, відібрана від усіх контрольованих одиниць упаковки.
Відбирають проби із пляшок, упакованих у ящики, із різних місць (ящиків) із розрахунку не менше однієї пляшки на 1 т олії, але не менш як чотири пляшки від кожної партії; перевіряють стан посуду, закоркування, етикетки, масу олії. Після перемішування вмісту відливають однакові порції олії в чистий сухий стакан для складання середньої проби об'ємом 2 дм3.
Середню пробу олії, розфасованої в бочки, фляги, бідони, відбирають трубчастим пробовідбірником із 10% одиниць упаковки, але не менш як із чотирьох одиниць. За наявності у партії олії менш як чотири одиниці упаковки пробу відбирають від кожної її одиниці.
Завдання 3. Дослідити якість рослинної олії за органолептичними і фізико-хімічними показниками.
Олія різних видів має специфічні органолептичні показники, які дають змогу визначити її вид (за винятком рафінованих дезодорованих олій, що не мають характерних властивостей) та ступінь її свіжості. Ступінь вираженості специфічних запаху, смаку і кольору олії залежить від виду сировини, способу її добування, ступеня очищення, умов і тривалості зберігання. Олія, одержана із пророслого, запліснявілого, підгорілого та іншого дефектного насіння, відрізняється неприємним смаком, запахом і темним кольором. Яскравий, золотисто-солом'яний колір означає наявність в олії каротиноїдів, а зеленуваті відтінки обумовлені хлорофілом. У разі порушення умов зберігання смак олії стає гострим, пекучим, згірклим.
Запах, колір і прозорість олії визначають при температурі 20°С (згідно з ГОСТ 5472-50 «Масла растительные. Определение запаха, цвета и прозрачности»).
Прилади і матеріали: стакан В-1-100 або В-1-150 за ГОСТ 25336-82, циліндр 2-100 або 4-100 за ГОСТ 1770-74, термометр рідинний скляний за ГОСТ 28498-90, баня водяна, пластинка скляна розміром 10-30 см.
Для визначення запаху олію наносять тонким шаром на скляну пластинку або розтирають на зворотному боці руки. Для кращого розпізнавання запаху олію нагрівають на водяній бані до 50°С.
Для визначення кольору олію наливають у хімічний стакан шаром не менш як 50 мм і розглядають на білому тлі у відбитому світлі або у світлі, що проходить.
Прозорість визначають таким чином: 100 см3 олії наливають у циліндр і залишають у спокої при температурі 20°С на 24 год (касторову олію - на 48 год). Відстояну олію розглядають як у відбитому світлі, так і у світлі, що проходить, на білому тлі. Олія прозора, якщо вона не має помутніння або завислих пластівців (бавовняна олія вважається прозорою, якщо вона не має помутніння або завислих пластівців у верхній частині циліндра).
Визначення колірного числа. Колірне число характеризує інтенсивність забарвлення олії, зумовлене наявністю в ній жиророзчинних пігментів, та глибину її очищення. Темне забарвлення погіршує органолептичні властивості олій. Величина нормується стандартами. Нерафіновані олії є темніші за рафіновані, тобто їх колірні числа є вищі. Колірне число та кислотне число є найбільш характерними показниками у визначенні виду (залежно від ступеня очищення) і товарного сорту олії.
Метод визначення заснований на порівнянні інтенсивності забарвлення досліджуваної олії із забарвленням стандартних розчинів (еталонів) йоду.
Колірне число виражається кількістю міліграмів вільного йоду, який міститься у 100 см3 його стандартного розчину, і має при однаковій з олією товщині шару (1 см) таку саму інтенсивність забарвлення, як і досліджувана олія.
Колірне число визначають за шкалою стандартних розчинів йоду (для бсіх олій, які мають у натуральному вигляді жовтий колір різної інтенсивності).
Прилади, обладнання: пробірка із безколірного скла із внутрішнім діаметром 10 мм; стакани з притертою пробкою; мірні колби місткістю 100 та 250 см3; градуйована піпетка місткістю 10 см3; бюретка із відтягнутим кінцем.
Реактиви: стандартний розчин йоду. Для його приготування у стакані з притертою пробкою зважують 0, 26-0, 27 г йоду і подвійну кількість КІ та розчиняють у 10 см3 дистильованої води.
Розчин переносять у мірну колбу на 250 см3 та при змішуванні доливають дистильовану воду до позначки і струшують. Концентрацію одержаного розчину визначають титруванням 0, 01 моль/дм2 розчином сіркуватокислого натрію (гіпосульфітом).
До приготовленого розчину додають, розраховуючи, дистильовану воду в такій кількості, щоб стандартний розчин містив точно 100 мг йоду у 100 мл.
Для приготування колірної шкали стандартного розчину йоду у пробірки (попередньо прокип'ячені з 10%-м розчином соляної кислоти і ретельно промиті) наливають піпеткою стандартний розчин йоду і додають із бюретки дистильовану воду в кількості, що наведена у таблиці:
| № пробірки | Стандартний розчин йоду, мл | Дистильованна вода, мл | № пробірки | Стандартний розчин йоду, мл | Дистильована вода, мл | |||||||
| - | 3, 5 | 6, 5 | ||||||||||
| 3, 0 | 7, 0 | |||||||||||
| 2, 5 | 7, 5 | |||||||||||
Закінчення таблиці
| № пробірки | Стандартний розчин йоду, мл | Дистильована вода, мл | № пробірки | Стандартний розчин йоду, мл | Дистильована вода, мл |
| 2, 0 | 8, 0 | ||||
| 1, 5 | 8, 5 | ||||
| 1, 2 | 8, 8 | ||||
| 4, 5 | 5, 5 | 1, 0 | 9, 0 | ||
| 4, 0 | 6, 0 | 0, 5 | 9, 5 | ||
| 0, 1 | 9, 9 |
Проведення випробування. У пробірку з безколірного скла із внутрішнім діаметром 10 мм наливають профільтровану олію і порівнюють інтенсивність її забарвлення із забарвленням стандартних розчинів йоду.
Випробування провадять при денному світлі, яке проходить, або при світлі матової електричної лампочки.
Колірне число випробуваної олії приймають таким, що дорівнює колірному числу еталона, і має однакове забарвлення з олією.
Колірне число приготовлених стандартних розчинів (еталонів) таке: у першій пробірці - 100, у другій - 90, у третій - 80, у четвертій - 70, у п'ятій - 60, у шостій - 50, у сьомій - 45, у восьмій - 40, у дев'ятій - 35, у десятій - 30, в одинадцятій - 25, у дванадцятій - 20, у тринадцятій - 15, у чотирнадцятій - 12, у п'ятнадцятій - 10, у шістнадцятій - 5, у сімнадцятій - 1.
Визначення кислотного числа. Кислотне число показує кількісний вміст у жирі вільних жирних кислот, накопичення яких зумовлено гідролітичним розщепленням гліцеридів на дигліцериди, моногліцериди, гліцерин та жирні кислоти.
Частково вільні жирні кислоти утворюються і внаслідок окислювальних перетворень жиру на більш пізніх стадіях його окислення.
За кількістю вільних жирних кислот, що містяться в жирі, можна судити про його свіжість, тому що у природних жирах їх мало. При неправильному зберіганні кількість вільних жирних кислот зростає і подальше їх окислення призводить до появи дефектів смаку і запаху, а при більш глибоких процесах - до непридатності жиру для харчових цілей. Тому кислотне число є одним із основних хімічних показників, згідно з яким олії, як і інші жири, поділяють на товарні сорти.
Значення кислотного числа для деяких олій наведено в табл. 6.1.
Таблиця б. 1
| Вид | ||||||||||||||||||
| Рафінована | Гідратована | Нерафінована | ||||||||||||||||
| Олія | Сорт | Сорт | ||||||||||||||||
| дезодорована | недезо- дорована | вищий | перший | ДРУ- гии | вищий | перший | другий | |||||||||||
| Соняшникова | 0, 4 | 0, 4 | 1, 5 | 2, 25 | 6, 0 | 1, 5 | 2, 25 | 6, 0 | ||||||||||
| Кукурудзяна | 0.4 | 0, 4 | - | - | - | 5, 0 | Сортів немає | |||||||||||
| Соєва | 0, 3 | - | - | 1, 0 | 1, 5 | - | 2, 0 | 4, 0 | ||||||||||
| Льняна | 0, 7 | - | - | - | - | - | 2, 5 | 5, 0 | ||||||||||
| Гірчична | - | - | - | - | - | 1, 5 | 2.3 | 6, 0 | ||||||||||
| Арахісова | 0.4 | 0.4 | 1, 5 | 2.25 | 6, 0 | 1, 5 | 2, 25 | 6, 0 | ||||||||||
Кислотне число жиру визначається кількістю міліграмів гідрооксиду калію або натрію, які потрібні для нейтралізації вільних жирних кислот, що містяться в 1 г жиру.
Кислотне число визначають згідно з ГОСТ 5476-80 «Масла растительные. Метод определения кислотного числа».
Сутність методу полягає в розчиненні певної маси рослинної олії в суміші розчинників із подальшим титруванням вільних жирних кислот спиртовим розчином гідрооксиду калію або натрію.
Прилади та обладнання: конічні колби місткістю 150-200 см3 за ГОСТ 25336-82, бюретка місткістю 50 см3 за ГОСТ 20292-74, водяна баня.
Реактиви: 1%-й спиртовий розчин фенолфталеїну або тимолфталеїну; спиртовий розчин гідрооксиду калію або натрію концентрації 0, 1 моль/дм3; нейтральна суміш спирту та ефіру.
Проведення випробування. У конічну колбу зважують з похибкою не більш за 0, 01 г 3-5 г олії, доливають 50 см3 нейтралізованої суміші розчинників і струшують. Якщо при цьому олія не розчиняється, її нагрівають на водяній бані, а потім охолоджують до температури 15...20°С. Одержаний розчин олії при постійному струшуванні швидко титрують розчином гідрооксиду калію або натрію концентрації 0, 1 моль/дм3 до одержання слабо-рожевого забарвлення.
За кінцевий результат приймають середнє арифметичне двох паралельних випробувань.
Визначення нежироеих домішок (відстою за масою). Нежирові домішки - це осад речовин різної хімічної природи, нерозчинних у петролейному ефірі або бензині. До таких речовин у рослинних оліях належать фосфатиди, воски, стерини, а також комплексні сполуки цих речовин. У рафінованих та гідратованих оліях нежирові домішки відповідно до чинних стандартів не допускаються. В оліях нерафінованих їх вміст є невеликий (залежно від сорту: 0, 05-0, 2%) і характеризує ступінь очищення олій. Сутність методу полягає у визначенні об'єму осаду, що утворюється в олії після відстоювання.
Прилади, обладнання: стакан хімічний місткістю 400 см3, складні мірні циліндри місткістю 100 см3 з поділками 0, 5 см3, термометр на 100°С, баня водяна.
Проведення випробування. 120 г олії нагрівають на водяній бані до 50°С, потім повільно охолоджують до 20°С та перемішують. У мірний циліндр наливають 100 см3 олії та залишають у спокої на 24 год при температурі 15...20°С, після чого відраховують об'єм осаду.
Об'єм осаду у мілілітрах є процентом відстою за об'ємом.
Визначення масової частки фосфоровміщуючих речовин.
Визначення здійснюється ваговим і колориметричним методами. Ваговий метод застосовується в межах 0, 05-6, 0% в перерахунку на стеароомолецитин і 0, 04-0, 53% в перерахунку на Р205. Колориметричний метод застосовується в межах 0, 006-6, 0% в перерахунку на стеароомолецитин і 0, 0005-0, 53% в перерахунку на Р205.
Сутність методу полягає в сухому спалюванні олії з окисом магнію (М^О) і подальшому визначенні фосфоровміщуючих речовин ваговим або калориметричним методами.
Прилади і матеріали. Ваги лабораторні за ГОСТ 24104-88, класу точності 2, з граничним навантаженням 200 г; електроплита побутова за ГОСТ 14919-83; насос водоструменевий лабораторний скляний за ГОСТ 25336-82, який забезпечує остаточний тиск не більше 2к Па (15 мм рт.ст.); шафа сушильна лабораторна; піч муфельна електрична з температурою 800-1000°С; щипці муфельні для чашок; ексікатор за ГОСТ 25336-82; тігель ТФ-20 ПФ 40 ХС - за ГОСТ 25336-82; чашки фарфорові для випаровування за ГОСТ 9147-80, № 2, діаметром 75 мм або чашки з прозорого кварцевого скла за ГОСТ 19908-80, діаметром 67 мм і більше; стакани хімічні за ГОСТ 25336- 82, ємністю 100 і 150 см3; циліндри мірні за ГОСТ 1770-74, ємністю 10, 25 і 50 см3; палички скляні; окис магнію за ГОСТ 4526-75, ч.д.а, свіжопрокалена; кислота азотна за ГОСТ 4461-77, густиною 1, 37-1, 40 і 1, 2 г/см; кислота сірчана за ГОСТ 4204-77, х.ч. або ч.д.а., густиною 1, 835 г/см3.
Реактиви: амоній сірчанокислий за ГОСТ 3769-78; амоній азотнокислий за ГОСТ 22867-77, 2%-й розчин; амоній молібденовокислий за ГОСТ 3765-78; ацетон за ГОСТ 2603-79, х.ч. або спирт етиловий технічний за ГОСТ 17299-78; спирт етиловий ректифікований технічний за ГОСТ 18300-87 і ефір диетиловий; вода дистильована з ГОСТ 6709-72; папір фільтрувальний за ГОСТ 12026-76.
Підготовка до випробування. 50 г сірчанокислого амонію зважують з похибкою не більше 0, 01г, розчиняють у 450 см ' концентрованої азотної кислоти густиною 1, 4 г/см3.
150 г молібденовокислого амонію зважують з похибкою не більше 0, 01 г, розчиняють в 400 см3 гарячої води (65-75°С), охолоджують до кімнатної температури та вливають при перемішуванні в розчин сірчанокислого амонію та азотної кислоти. Доводять об'єм водою до 1 дм3 і ставлять у темне місце на 2 доби, після чого розчин фільтрують та зберігають у склянці з темного скла. Густина розчину 1, 315-1, 320 г/см3.
Приготування суміші азотної і сірчаної кислот.
1 дм3 азотної кислоти густиною 1, 20 г/см3 змішують з 30 см3 концентрованої сірчаної кислоти густиною 1, 835 г/см3.
Підкислення розчину азотнокислого амонію.
До 1 дм3 2%-го розчину азотнокислого амонію добавляють 5 см3 20%-ї або 4-5 крапель концентрованої азотної кислоти.
Перевірка окису магнію.
Окис магнію не повинен містити солей фосфорної кислоти. Для перевірки окису магнію на чистоту при проведенні кожної серії аналізів олії роблять контрольне спалювання 2, 5 г окису магнію, зваженого з похибкою не більше 0, 01 г, без олії з подальшою обробкою прокаленого препарату реактивами як при проведенні випробування з олією (див. далі «Проведення випробування»).
Підготовка ацетону.
Ацетон зневоднюють хлористим кальцієм або оцтово-кислим калієм, нейтралізують по фенолфталеїну або лакмусу та переганяють при температурі не вище 60°С.
Підготовка фільтруючого тигля.
Тигель промивають два рази ацетоном або послідовно два рази спиртом і два рази диетиловим ефіром, висушують протягом 1 год у вакуум-ексікаторі або в сушильній шафі при температурі 100°С до постійної маси і зважують з похибкою не більше 0, 0001 г.
Підготовка проби.
Пробу досліджуваної олії добре перемішують, нагрівають до температури 70-75°С і фільтрують при цій самій температурі.
Проведення випробування. У фарфорову чашку зважують 2, 0-2, 5 г олії з точністю до 0, 0001 г. Додають 2, 5 г окису магнію (зважену з точністю до 0, 01 г) і нагрівають у сушильній шафі при температурі 110°С близько 10 хв для того, щоб олія адсорбувалася окисом магнію, потім нагрівають на електроплиті до обвуглювання і залишок прокалюють до білого кольору у муфельній печі при 800-1000°С протягом 1 год.
Охолоджують муфель до 400-300°С. Виймають чашки і охолоджують їх до кімнатної температури, осад переносять у хімічний стакан і обережно по стінках приливають 10 см3 води. Осад, що залишився в чашці, змочують декількома краплями води, приливають 15 см^ приготовленої суміші азотної і сірчаної кислот і, перемішуючи паличкою, розчиняють осад при слабкому перемішуванні.
Вміст чашки переносять у стакан, промивають двічі чашку при слабкому нагріванні 10 мл суміші кислот (по 10 см3). При використанні кварцевих чаш діаметром 95 і 105 мм допускається розчиняти осад сумішшю кислот і осаджувати осад за допомогою молібде- новокислотго амонію безпосередньо в чашах. Отриманий розчин нагрівають до кипіння, знімають з плитки і охолоджують на повітрі до 50°С. Потім приливають 50 см3 розчину молібденовокислого амонію. Перемішують вміст обертанням стакана або чаші, залишають рідину на 2 год для формування осаду. Потім вміст стакану декантують через фільтруючий тигель, змиваючи осад зі стакану 2%-м розчином азотнокислого амонію.
Переносять осад на фільтр, промивають два рази ацетоном (по 10-15 см3) або послідовно два рази спиртом (по 10-15 см3) і два рази диетиловим спиртом (по 10-15 см3).
Осад висушують у сушильній шафі при температурі 100°С і доводять до постійної маси. Зважують через 30 хв з похибкою не більше 0, 0001 г.
Масову частку окису фосфору у нерафінованих і гідратованих оліях (X) у процентах розраховують за формулою:
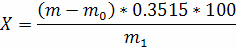
де m0 - маса осаду в контрольному досліді, г;
m - маса осаду в основному досліді, г;
0, 03515 - коефіцієнт, який знайдено, виходячи із вмісту окису фосфору в осаді фосфорномолібденового амонію;
M1 - маса наважки, г.
Результат випробування округляють до другого десяткового знака для величини більше 0, 01% при розрахунку на стеаролецитин і до третього десяткового знака для величин більше 0, 0001% при перерахунку на Р205.
Масову частку фосфоровміщуючих речовин в оліях у процентах у перерахунку на стеаролеолецитин розраховують за формулою
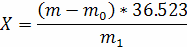
Деm0 - маса осаду в контрольному досліді, г;
m - маса осаду в основному досліді, г;
36, 523 - коефіцієнт для перерахунку маси осаду комплексної солі фосфорномолібденового кислого амонію на стеароолео- лецитин;
m1 - маса наважки, г.
Для рафінованих олій результат випробування менше 0, 05% приймають за відсутність фосфоровміщуючих речовин.
Визначення вологи та летких речовин (ГОСТ11812-66).
Прилади і матеріали: стаканчики для зважування за ГОСТ 25336-82 або бюкси алюмінієві діаметром 50 мм та висотою 50 мм; ексікатор за ГОСТ 25336-82; шафа сушильна лабораторна з терморегулятором; ваги лабораторні за ГОСТ 24104-88 2-го класу точності з найменшою межею зважування 200 г.
Проведення випробування. У попередньо висушений (при температурі 100-105°С протягом 30 хв) до постійної маси стаканчик зважують 5 г досліджуваної олії (результат до четвертого знака після коми) і висушують її при температурі 100-105°С до постійної маси.
Перше зважування здійснюють після висушування олії протягом 20 хв, наступні - після 15 хв висушування. Постійна маса вважається досягнутою, якщо зменшення маси при двох послідовних зважуваннях не перевищує 0, 0005 г.
Масову частку вологи та летких речовин у досліджуваній олії (X) у процентах розраховують за формулою
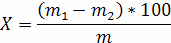
де m - маса олії, яка досліджується, г;
m1- маса стаканчика з олією до висушування, г;
m2 - маса стаканчика з олією після висушування, г.
Кінцевий результат випробування виражається як середнє арифметичне з двох паралельних визначень. Допустимі розходження між паралельними випробуваннями не повинні перевищувати 0, 04%.
Визначення мила (якісна проба) — ГОСТ 5480-59.
Цим методом визначається відсутність мила (натрієвих солей вільних жирних кислот) у рафінованих оліях після лугової рафінації.
Прилади і матеріали. Плитка електрична; азбест; скляні кульки або шматочки пемзи, або скляні капіляри; колби Кн (Пі) - 250 Тс за ГОСТ 25336-82; циліндри 1(3) - 25 (50, 100) за ГОСТ 1770-74; вода дистильована за ГОСТ 6709-72.
Реактиви. Фенолфталеїн за ТУ 6-09-536-87, спиртовий розчин з масовою часткою 1%; спирт етиловий за ГОСТ 17299-78 або за ГОСТ 18300-87.
Проведення випробування. Для проведення випробування беруть ретельно перемішану непрофільтровану олію. У конічній колбі ємністю 250 см3 попередньо кип'ятять 50 см3 дистильованої води з декількома краплями фенолфталеїну (при цьому вода повинна залишитися безколірною), потім додають близько 10 см3 досліджуваної олії і кип'ятять протягом 5-10 хв.
Для рівномірності кипіння в колбу поміщають шматочки пемзи або скляні капіляри.
Після закінчення кип'ятіння колбу ставлять на аркуш білого паперу і додають ще декілька крапель фенолфталеїну.
При відсутності мила в олії нижній шар у колбі після охолодження повинен залишитися безколірним. Чутливість методу 0, 020%.
Визначення температури спалаху екстракційної олії. Сутність методу полягає в тому, що леткі продукти і продукти розпаду компонентів олії, які в ній містяться, при нагріванні в певних умовах утворюють з повітрям суміш, що спалахує при піднесенні полум'я. Значною мірою на величину температури спалаху впливає вміст у жирах вільних жирних кислот. Тому жири з підвищеним кислотним числом мають нижчу температуру спалаху, що значно впливає на ретельність видалення залишків бензину, який знижує температуру спалаху.
Прилади і матеріали. Прилад типу ПВНЗ з електричним нагрівом або типу ПВНО з полум'яним (газовим або бензиновим) нагрівом, який забезпечує визначення температури спалаху в інтервалі 150-250°С, швидкість переміщення зразка 60 об/хв і швидкість нагріву 2°С/хв; секундомір, екрануючі щити з покривної сталі 450x600 мм, сірники за ГОСТ 1820-85.
Проведення випробування. Олію, яку досліджують, наливають у тигель до кругового уступу, закривають чистою сухою кришкою, вставляють термометр і поміщають тигель у нагрівальну ванну. Запалюють гніт лампочки, попередньо налиту рафінованою олією, або газовий пальник, і регулюють полум'я таким чином, щоб його форма наближалася до форми кулі діаметром 3-4 мм. Для кращого захисту від руху повітря та впливу світла прилад оточують/
Повноту відмивання клейковини можна перевірити таким чином:
до краплі води, віджатої з відмитої клейковини, додають краплю розчину йоду в йодистому калію - відсутність синього забарвлення свідчить про повне видалення крохмалю;
в чисту воду, налиту у вимитий стакан, віджимають із клейковини 2-3 краплі промивної води. Крохмаль у клейковині відсутній повністю, якщо немає муті.
Підмиту клейковину добре віджимають руками, витираючи їх чиї від часу сухим рушником. Віджату клейковину зважують на н мнчпих вагах з точністю до 0, 01 г.
Пісня першого зважування клейковину ще раз промивають інірпдоиж 5 хв під струменем води, знову віджимають і зважують. Її шипи між двома зважуваннями не має перевищувати 0, 1 г.
При визначенні температури спалаху соєвої олії в разі підвищеного вмісту в ній фосфатидів і вологи, для запобігання спінення олії при нагріванні перед випробуванням її центрифугують протягом 5-10 хв зі швидкістю 1000-1500 об/хв" 1.
Потім прилад нагрівають на газовому, бензиновому або спиртовому пальнику або електрикою. Протягом всього періоду нагрівання олію безперервно перемішують пружинною мішалкою з частотою обертання 1 с'1 (60 об/хв). Тільки в момент випробування на спалах перемішування припиняють. Спочатку нагрівають на великому полум'ї, щоб температура досягла 170°С протягом 15-20 хв, після чого нагрівання уповільнюють. Коли олія нагрівається до температури на 30°С нижче очікуваної температури спалаху, нагрівання продовжують таким чином, щоб температура досліджуваної олії підвищувалася зі швидкістю 2°С за хвилину. При температурі на 10°С нижче очікуваної температури спалаху починають проводити випробування на спалах, відкриваючи прилад поворотом пружинного важеля точно через хвилину, тобто через кожні 2°С (час спостерігають за секундною стрілкою годинника). Перед підпалюванням перемішування припиняють і відкривають отвір кришки на 2-3 с. Випробування на спалах можна також проводити за допомогою попередньо запаленого сірника, опускаючи його полум'я в паровий простір. Якщо спалах не відбувся, олію знов перемішують, повторюючи спалах через хвилину.
Моментом спалаху вважається момент появи синього полум'я. Після першого спалаху випробування продовжують, повторюючи підпалювання через хвилину. Якщо при цьому спалаху не буде, випробування повторюють знову. Якщо при новому випробуванні температура спалаху, одержана при першому визначенні, повториться, а повторного спалаху через 2°С також не відбудеться, випробування вважають закінченим і за температуру спалаху приймають показання термометра в момент першої появи синього полум'я над поверхнею досліджуваної олії при двох паралельних випробуваннях. Після випробування олію зливають з тигля, тигель з кришкою промивають водою будь-який мийним засобом і ретельно висушують.
Допустимі розбіжності між двома паралельними випробуваннями зі свіжими порціями олії не повинні перевищувати 3°С.
При більшій розбіжності між двома паралельними випробуваннями роблять третє випробування зі свіжою олією і за кінцевий результат беруть середнє арифметичне з двох визначень, які відрізняються один від одного не більше ніж на 3°С.
Визначення ступеня прозорості соняшникової олії
Апаратура: фотоколориметр, який дозволяє проводити вимірювання при довжині хвиль 570 або 590 нм; ваги лабораторні загального призначення за ГОСТ 24104-88, 2-го класу точності і найбільшою межею зважування 200 г або інші ваги з аналогічними характеристиками; шафа електрична сушильна з терморегулятором, який забезпечує похибку підтримки температури не більше 2°С; термометри лабораторні типу ТД-2 за ГОСТ 28498-90; стакани хімічні за ГОСТ 1770-74; колби конічні за ГОСТ 25336-82; піпетки за ГОСТ 20292-74; бюретки за ГОСТ 20292-74.
Реактиви і матеріали: гідразин сірчанокислий за ГОСТ 5841- 74; уротропін технічний за ГОСТ 1381-73 або фармакопейний для ін'єкцій; спирт етиловий ректифікований технічний за ГОСТ 18300- 87; вода дистильована за ГОСТ 6709-72; папір фільтрувальний за ГОСТ 120026-76.
Підготовка до випробування. Прилад (фотоколориметр) готують відповідно до інструкції по його експлуатації. Установлюють фільтр з довжиною хвиль 570 або 590 нм.
Приготування суспензій формазину. За одиницю формазинової шкали приймають розведену у співвідношенні 1: 1000 водну суспензію формазину, яку одержану при взаємодії однакових об'ємів водного розчину сірчанокислого гідразину масової концентрації 10 г/дм3 і водного розчину уротропіну масової концентрації 100 г/дм3.
Приготування розчину сірчанокислого гідразину масової концентрації 10 г/дм3. Розчини сірчанокислого гідразину готують у мірній колбі місткістю 100 см3. Зважують (1, 00+0, 01) г сірчанокислого гідразину. Результат записують з точністю до другого десяткового знака. Потім поміщають у мірну колбу. Приливають дистильовану воду, перемішують, доводять об'єм дистильованої води до мітки і знову ретельно перемішують. Розчин сірчанокислого гідразину перед приготуванням формазинової емульсії повинен стояти не менше 4 год.
Приготування водного розчину уротропіну масової концентрації 100 г/дм3. Розчин уротропіну готують у мірній колбі ємністю 100 см3. Зважують (10, 00+0, 01) г уротропіну. Результат записують з точністю до другого десяткового знака. Наважку поміщають у мірну колбу. Приливають дистильовану воду, перемішують, доводять об'єм дистильованої води до мітки і знову ретельно перемішують.
Приготування вихідної суспензії зі ступенем прозорості 1000 формазинових одиниць (фем). Водні розчини сірчанокислого гідразину та уротропіну з мірних колб вливають у конічну колбу ємкістю 250 см3, ретельно перемішують. Суміш залишають на 24 год при температурі (20+2)°С для одержання стійкої суспензії.
Приготування суспензії формазину зі ступенем прозорості 50 фем. У мірну колбу ємкістю 500 см3 піпеткою вводять 25 см3 вихідної суспензії, доводять об'єм дистильованої води до мітки і перемішують.
Приготування суспензії формазину зі ступенем прозорості 2 фем. У мірну колбу ємкістю 500 см3 піпеткою вводять 1 см3 вихідної суспензії формазину, доводять об'єм до мітки дистильованою водою і перемішують.
Перед відбором необхідної кількості необхідно ретельно перемішувати суспензії формазину для рівномірного розподілу частинок по об'єму колби.
Градуювальні суспензії формазину зберігають у колбах з притертими пробками у холодильнику при температурі 5-10°С. Суспензії формазину зберігають стабільними значення оптичної густини: зі ступенем прозорості 50 фем протягом 1 міс; зі ступенем прозорості 2 фем протягом 5 діб.
Побудова градуювального графіка. Градуювальний графік будують, використовуючи дві градуювальні суспензії зі значенням ступеня прозорості 2 і 50 фем. Для побудови градуювального графіка заливають в одну кювету фотоколориметра дистильовану воду, а в дві інші - градуювальні суспензії зі ступенем прозорості 2 і 50 фем. Кювету з дистильованою водою почергово поміщають з відповідною градуювального суспензією (2 або 50 фем) в обидва вимірювальних канали приладу і записують значення оптичної густини для кожної суспензії. За отриманими двома значеннями оптичної густини будують лінійний графік: ступінь прозорості - оптична густина.
Підготовка проби олії. Якщо олія була охолоджена, її підігрівають до кімнатної температури, перемішують, наливають 50-60 см3 олії в стакан, підігрівають у сушильній шафі до 80-85°С і фільтрують безпосередньо в сушильній шафі через складчастий фільтр для відділення нежирових домішок, розміщуючи пробу біля чутливого елемента термометра. Профільтровану олію охолоджують до температури 20-22°С у проточній воді.
Проведення випробування. Підготовлену олію наливають, щоб не утворилися пухирці повітря, в кювету фотоколориметра довжиною 20 мм; кювету швидко поміщають у прилад і вимірюють оптичну густину олії відносно кювети з тією самою олією, але профільтрованою через складчастий фільтр при температурі (20+2)°С. Результат записують з точністю до першого десяткового знака. Вимірювання проводять компенсаційним методом. Вимірявши оптичну густину досліджуваної олії, за градуювальним графіком визначають ступінь його прозорості у формазинових одиницях (фем). Результат записують з точністю до першого десяткового знака. За кінцевий результат випробування приймають середнє арифметичне значення ступеня прозорості двох зразків підготовленої олії.
Визначення перекисного числа. Перекисне число свідчить про наявність в оліях первинних продуктів окиснення жирів - перекисів. Тобто, це показник, який свідчить про свіжість олій. Значення цього показника збільшується в процесі зберігання.
Метод визначення перекисного числа базується на окисненні йодистого калію перекисами і гідроперекисами, які містяться в зразку олії, розчині оцтової кислоти та хлороформі і титруванням йоду, що виділився, розчином тіосульфату натрію.
Прилади і матеріали: ваги лабораторні за ГОСТ 24104-80, класу точності 2 з найбільшою межею зважування 200 г або інші ваги такого самого класу точності; колби за ГОСТ 25336-82 ємкістю 250 см3; стаканчики скляні циліндричні, бюретки за ГОСТ 20292-74 ємністю 5 і 10 см3.
Реактиви: хлороформ за ГОСТ 20016-74 свіжоперегнаний; кислота оцтова льодяна за ГОСТ 61-75, х.ч., калій йодистий за ГОСТ 4232-74, х.ч., 50-55%-й розчин свіжоприготовлений; натрій сіркова- тистокислий (тіосульфат натрію), водний розчин 0, 01 моль/дм3 (0, 01 н) або 0, 002 моль/дм (0, 02 н); крохмаль розчинний за ГОСТ 10163-76, 0, 5%-й розчин; вода дистильована.
Вимоги безпечності. Хлороформ - негорючий, має загальноток- сичну дію, його зберігають у герметичному посуді з темного скла. Оцтова кислота - легкозаймиста рідина зі специфічним запахом. Робота з хлороформом і оцтовою кислотою проводиться з дотриманням правил особистої гігієни і використанням витяжної шафи.
Категорично забороняється виливати хлороформ і оцтову кислоту в каналізацію.
Підготовка до випробування. Розчин йодистого калію зберігають у темному посуді. Перед використанням його перевіряють. Для цього доливають 2 краплі розчину крохмалю до 0, 5 см3 розчину йодистого калію в 30 см3 розчину оцтової кислоти і хлороформу (3: 2). Якщо утворюється блакитне забарвлення, для знебарвлення розчину потрібно більше 1 краплі 0, 01 моль/дм3 розчину тіосульфіту натрію, то розчин йодистого калію не використовують і готують свіжий розчин.
Розчин крохмалю готують таким чином: 5 г розчинного крохмалю змішують з 30 см3 води, додають цю суміш до 1000 см киплячої води і кип'ятять протягом З хв.
Проведення випробування. Масу проби, необхідну для випробування, залежно від можливого перекисного числа, визначають за табл. 6.2.
Таблиця 6.2
| Можливе значення перекисного числа, МІМОЛЬ/КГ | Маса проби, г | |||
| Від 0 до 6, 0 | 5, 0-2, 0 | |||
| Вище 6, 0 до 10, 0 | 2, 0-1, 2 | |||
| Вище 10, 0 до 15, 0 | 1, 2-0, 6 | |||
| Вище 15, 0 до 25, 0 | 0, 6-0, 5 | |||
| Вище 25, 0 до 40, 0 | 0, 5-0, 3 | |||
Пробу зважують у колбу. Додають 10 см3 хлороформу, швидко розчиняють пробу, додають 15 см3 оцтової кислоти і 1 см-' розчину йодистого калію, після чого колбу відразу закривають, перемішують вміст протягом 1 хв і залишають на 5 хв у темному місці при температурі 15-25°С. Потім додають 75 см^ води, ретельно перемішують і додають п'ять крапель розчину крохмалю. Іод, що виділився, титрують розчином тіосульфату натрію, використовуючи розчин такої концентрації залежно від можливого значення перекисного числа: С(1/2На2$20з) = 0, 02 моль/дм3, якщо перекисне число не більше 6, 0 ммоль/кг або з (1/2№28203) = 0, 01 моль/дм3, якщо перекисне число дорівнює або більше 6, 0 ммоль/кг.
Для кожної досліджуваної проби виконують два вимірювання.
Контрольне вимірювання (без наважки олії) проводять паралельно з основними вимірюваннями (з наважкою олії).
Якщо на контрольне вимірювання піде більше 0, 1 см3 0, 01 моль/дм3 розчину тіосульфату натрію, то перевіряють відповідність реактивів вимогам стандарту.
Перекисне число (X) у мілімолях активного кисню на кілограм проби розраховують за формулою:
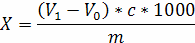
де V0 - об'єм розчину тіосульфату натрію, який використаний при контрольному вимірюванні, см3;
V1 - об'єм розчину тіосульфату натрію, який використаний при основному вимірюванні, см3;
с - концентрація використаного розчину тіосульфату натрію,
ммоль/дм3;
m - маса досліджуваної проби, г;
1000 - коефіцієнт, який враховує перерахунок результату вимірювання у мілімолі на кілограм.
За результат вимірювання приймають середнє арифметичне значення двох паралельних вимірювань.
Визначення показника заломлення. Жир заломлює світло різного ступеня залежно від складу і структури. За величиною показника заломлення можна судити про природу олії, її чистоту і ступінь окислення. Здатність жирів до заломлення зростає при збільшенні молекулярної маси, а також ненасиченості жирних кислот. Показник заломлення різних видів олій наведено в табл. 6.3.
Таблиця 6.3
| Вид олії | Показник заломлення при температурі 20°С | Вид олії та жиру | Показник заломлення при температурі 20°С |
| Соняшникова | 1, 4736-1, 4762 | Масло какао | 1, 4530-1, 4580 |
| Соєва | 1, 4722-1, 4754 | Баранячий жир | 1, 4383-1, 4566 |
| Бавовняна | 1, 4742-1, 4768 | Яловичий жир | 1, 4510-1, 4583 |
| Арахісова | 1, 4680-1, 4720 | Свинячий жир | 1, 4536-1, 4588 |
| Льняна | 1, 4858-1, 4872 | ||
| Конопляна | 1, 4717-1, 4780 | ||
| Кукурудзяна | 1, 4720-1, 4740 | ||
| Гірчична | 1, 4730-1, 4769 |
В окислених жирах числове значення показника заломлення є вищим порівняно зі свіжими внаслідок збільшення молекулярної маси (приєднання кисню, окис груп тощо).
Показник заломлення визначають згідно з ГОСТ 5482-90 «Масла растительные. Метод определения показателя преломления».
Світло, переходячи з менш густого середовища - повітря у більш густе - жир, потрапляє на поверхню їх розподілу під гострим кутом і заломлюється. При цьому напрям руху змінюється таким чином, що кут заломлення є меншим за кут падіння. Величина відношення
є постійною у даних умовах і характеризує величину показника заломлення. Показник заломлення залежить від температури, довжини хвилі Я світла, що падає, і позначається пВї. При зміні температури змінюється густина речовини: при підвищенні на 1°С густина знижується на 0, 00035 і, як наслідок, показник заломлення зменшується.
Прилади і матеріали: універсальний рефрактометр РЛУ, скляна паличка, вата або бинт.
Спочатку перевіряють рефрактометр, визначаючи коефіцієнт заломлення дистильованої води. Показник заломлення дистильованої води при температурі 10°С - 1, 3335; 15°С - 1, 3333; 20°С - 1, 3329; 30°С- 1, 3320.
Перевірку рефрактометра і випробування олії провадять при денному світлі або при світлі електричної лампочки. На нижню призму рефрактометра, не торкаючись її, скляною паличкою наносять декілька крапель олії, яка випробовується. Нижню призму присувають до верхньої, закріплюють за допомогою запору, ставлять дзеркало і окуляр так, щоб у полі зору був чітко видний перетин ниток.
Алідаду повільно рухають доти, поки межа затемненої частини поля зору не наблизиться до місця перетину ниток. При обертанні маховичка компенсатора дисперсії встановлюють різку межу між темною і світлою частинами поля зору, яку підводять точно в точку перетину ниток.
Після підведення межі затемненої частини поля зору точно в точку перетину ниток відраховують за шкалою приладу показник заломлення. Відрахунок виконують 2-3 рази з точністю до 0, 0002 після закінчення 5 хв з моменту встановлення певної температури і обчислюють середнє значення одержаних величин.
Якщо показник заломлення визначений не при 20°С, то його приведення до цієї температури обчислюють
Олію видаляють із поверхні призми сухою ватою, потім ватою, змоченою ефіром, та сухою м'якою тканиною.
Визначення йодного числа. Йодне число жиру виражають кількістю грамів йоду (або іншого галоїду у перерахунку на йод), яка може приєднатися до 100 г жиру1. Воно залежить від природи жиру та його свіжості. Чим більше у жирі є ненасичених жирних кислот, тим вищим є його йодне число, тому що при зростанні кількості подвійних (етиленових) зв'язків у вуглецево-водневому ланцюгу жирних кислот зростає кількість йоду, що приєднався. Рослинні олії внаслідок більшого вмісту ненасичених жирних кислот порівняно з тваринними жирами мають більш високі значення йодних чисел (табл. 6.4).
Таблиця 6.4
| Вид олії | Йодне число, % І2 | Вид жиру | Йодне число, % І2 | |||||
| Соняшникова | 125-145 | Баранячий | 31-46 | |||||
| Соєва | 120-140 | Яловичий | 32-57 | |||||
| Бавовняна | 101-116 | Свинячий | 46-54 | |||||
| Арахісова | 83-105 | Вершкове масло | 30-50 | |||||
| Льняна | 171-206 | |||||||
| Кукурудзяна | 111-133 | |||||||
1 Тобто йодне число характеризує ступінь його насиченості.
У процесі окислення жирів кількість ненасичених жирних кислот знижується і як наслідок зменшується йодне число.
Методи Гюбля, Гануса, Кауфмана, Війса, прискорений метод зі спиртовим розчином йоду, що використовуються, засновані на властивості ненасичених жирних кислот приєднувати галогени за місцем подвійних зв'язків. Жир обробляють надлишком спиртового розчину йоду (тому що повне насичення подвійних зв'язків відбувається лише тоді, коли кількість галоїду є на 50-60% вища за теоретичний) у присутності великої кількості води. За цих умов розчин йоду піддається гідролізу з утворенням йодисто-водневої та йоднуватистої кислот:
І2+Н20< н> НІ+НЮ.
Продукти дисоціації йоднуватистої кислоти (І+ОН) приєднуються за місцем подвійних зв'язків ненасичених жирних кислот
СН3...СН=СН...СООН+ШО= СНЗ...СНІ-СНОН...СООН.
У розчині залишається йодисто-воднева кислота НІ, кількість якої дорівнює кількості йоднуватистої кислоти НІО, витраченої на заміщення подвійних зв'язків жирних кислот. Непрореагована НІ відтитровується у робочому досліді (з наважкою жиру) розчином гіпосульфіту концентрації 0, 1 моль/дм3 у присутності індикатора крохмалю:
2Ма28203+І2=2КаІ +N328406.
Щоб дізнатися про кількість галоїду НІО, яка приєдналася до ненасичених жирних кислот досліджуваного жиру, слід провести в аналогічних умовах контрольний дослід (без наважки жиру). Різниця між кількістю гіпосульфіту, витраченою у контрольному і робочому дослідах, буде визначати ту кількість галоїду НІО, яка приєдналася до досліджуваного жиру.
Прилади та обладнання: колби із безколірного скла з пришліфованою пробкою за ГОСТ 25336-82 ємністю 500 см3, бюретка місткістю 25 і 50 см3 за ГОСТ 20292-74; піпетки ємністю 10, 15, 20 см3 за ГОСТ 20292-74, колби мірні ємністю 100 см3 за ГОСТ 1770-74, ваги лабораторні з найбільшою межею зважування 200 г, другого класу точності; водяна баня.
Реактиви: розчин гіпосульфіту концентрації 0, 1 моль/дм3, розчин йоду концентрації 0, 2 моль/дм3 (25 г йоду у 1000 см3 96%-го спирту), етиловий спирт, 1%-й розчин крохмалю.
Проведення випробування. Наважку олії масою 0, 2 г, зважену в колбі з похибкою до 0, 0002 г (взяту за різницею зважувань), розчиняють у 40 см3 спирту або при нагріванні на водяній бані при температурі 50°С (для повного розчинення жиру). Охолоджують, додають 25 см3 спиртового розчину йоду концентрації 0, 2 моль/дм3 та добре перемішують. Доливають 200 см3 води, знову перемішують і залишають у сцокої в темному місці на 5 хв для приєднання йоду до подвійних зв'язків ненасичених жирних кислот. Через 5 хв розчин із залишком непрореагованого йоду титрують розчином гіпосульфіту концентрації 0, 1 моль/дм3. Наприкінці титрування (коли розчин у колбі стане жовтим) додають 0, 5 см3 1%-го розчину крохмалю та продовжують титрування до зникнення синього забарвлення. Паралельно в аналогічних умовах досліду проводять контрольне випробування (без наважки жиру).
Йодне число жиру визначають за формулою
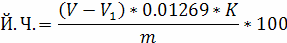
де V, Vі - відповідно об'єми розчину гіпосульфіту концентрації 0, 1 моль/дм3, витрачені на титрування контрольного та основного випробування, см3;
0, 01269-маса йоду, еквівалентна 1см3 розчину гіпосульфіту концентрації 0, 1 моль/дм3, г;
v- маса наважки жиру, г;
К - поправка до титру для перерахунку на розчин гіпосульфіту концентрації 0, 1 моль/дм3.
Після виконання всіх випробувань результати оцінки якості записують у табл. 6.5 та роблять висновок щодо якості олії.
Таблиця 6.5
| Показник якості | Характеристика показників олії згідно з ГОСТ 1129-93 | Дані лабораторного випробування | ||||||||
| Рафінована | Гідратована | Нерафінована | ||||||||
| дезодорована марки | неде- зодо- рована | вищого сорту | І сорту | II сорту | вищого сорту | І сорту | II сорту | |||
| Д | П | |||||||||
Контрольні питання
1. Характеристика насичених і ненасичених жирних кислот: їх фізичні та хімічні властивості.
2. Харчова цінність жирів залежно від їх жирнокислотного вмісту.
3. Речовини, супутні жирам: фосфатиди, воски, стерини, жиророзчинні вітаміни, барвні речовини. їх характеристика та вплив на харчову цінність, товарні властивості та зберігання жирів.
4. Процеси, що відбуваються у жирах під час зберігання. Антиокислювачі та синергісти: характеристика та сутність дії.
5. Порівняльна характеристика олій, одержаних пресуванням та екстракцією.
6. Методи очищення (рафінації) рослинних олій: сутність методів, їх вплив на харчову цінність та товарні властивості жирів.
7. Органолептичні та фізико-хімічні показники якості рослинних олій: сутність методик їх визначення.
8. Дефекти рослинних олій.
|