Главная страница Случайная страница
КАТЕГОРИИ:
АвтомобилиАстрономияБиологияГеографияДом и садДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеталлургияМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРелигияРиторикаСоциологияСпортСтроительствоТехнологияТуризмФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Метод функционала плотности
|
|
Наиболее эффективным, является метод расчета структуры и свойств молекул, который использовал бы только информацию о конфигурации электронных оболочек атомов, составляющих систему. Этот метода позволил бы ученым предсказывать существование и свойства новых материалов, еще не полученных экспериментально.
Первопринципныеметоды (аb-initio) используют этот механизм, но за счет большой сложности расчётов в них также применяются приближения. При этом существует высокая точность расчетов.
К основным методам первопринципных расчетов можно отнести следующие: метод Хартри-Фока и метод функционала электронной плотности.
Одним из самых первых методов является метод Хартри-Фока для расчета атомов и молекул, в связи с чем получил широкое распространение.
Однако при использовании этого метода, для больших систем возникают трудности из-за огромного количества перекрестных обменных интегралов. Кроме того, этот метод, учитывает обменное взаимодействие и не учитывает корреляционные эффекты, что делает его использование для расчётов электронной структуры некорректным, причем для поверхностей эти приближения подходят еще меньше, чем для объемных кристаллов.
В следствии этого в данной работе расчеты производились с использованием метода функционала электронной плотности (DFT).
В основе метода DFT лежит теорема Хоэнберга-Кона о том, что между электронной плотностью р в данной системе и ее электронной энергией существует однозначное соответствие, и электронная энергия основного состояния всецело определяется электронной плотностью. Применение метода функционала плотности непосредственно в вычислительной химии стало возможным с введением орбитального приближения Кона и Шэма.
Поскольку плотность ρ (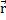 ) определяет как число частиц N, так и потенциал взаимодействия электронов с внешним полем Vee() (с точностью до несущественной аддитивной постоянной), с помощью нее можно получить полный гамильтониан H и оператор числа частиц N для электронной системы.
) определяет как число частиц N, так и потенциал взаимодействия электронов с внешним полем Vee() (с точностью до несущественной аддитивной постоянной), с помощью нее можно получить полный гамильтониан H и оператор числа частиц N для электронной системы.
Следовательно, ρ () неявно определяет свойства, получаемые из гамильтониана путем решения уравнения Шредингера зависящего или не зависящего от времени.
Таким образом, функционал полной энергии может быть записан в виде:
𝐸 [𝜌 (𝑟)] = 𝑇 [ρ ()]+ Uee[ρ ()] + 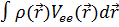 , (1)
, (1)
где 𝑇 [ρ ()] – учитывает кинетическую энергию электронов, Uee[ρ () ] - энергию межэлектронного взаимодействия, Vee() - потенциал, описывающий взаимодействие электронов с внешним полем. Минимизируя энергию относительно одноэлектронных орбиталей, можно получить уравнения Кона - Шема аналогичные уравнениям Хартри - Фока:
ℎ 𝑘 𝑠 𝜑 𝑖 = 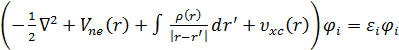 , (2)
, (2)
здесь hks - одноэлектронный оператор Кона-Шема, аналогичный оператору Фока, в уравнениях Хартри-Фока, ε i - энергия канонических одноэлектронных орбиталей Кона-Шема, 𝜑 𝑖 - канонические одноэлектронные орбитали Кона-Шема, Vne - потенциал электронно-ядерного взаимодействия и vxc - обменно-корреляционный потенциал.
Различные методы функционала плотности отличаются формой обменно-корреляционного функционала. Как правило, этот функционал выражается через плотность корреляционно-обменной энергии ε xc:
𝑉 𝑥 𝑐 𝑟 = 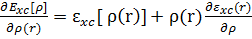 (3)
(3)
Корреляционно-обменная плотность энергии sxc может быть представлена как сумма корреляционной sc и обменной sx энергий.
Метод функционала плотности реализуется с помощью одного из трех приближения, от которых зависит вид обменно-корелляционного функционала.