Главная страница Случайная страница
КАТЕГОРИИ:
АвтомобилиАстрономияБиологияГеографияДом и садДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеталлургияМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРелигияРиторикаСоциологияСпортСтроительствоТехнологияТуризмФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Гидроформилирование алкенов (Оксосинтез)
|
|
Процесс гидроформилирования различных алкенов проводят для получения альдегидов и спиртов. Мировой объём производства оксопродуктов составлял примерно 8, 5 млн. т/г на начало 21-го века. Первичными продуктами оксосинтеза являются альдегиды, которые в разной степени в зависимости от используемой каталитической системы и условий гидрируются в спирты. Примерно 70% объема производства продуктов гидроформилирования составляют продукты превращения пропилена – масляные альдегиды и бутиловые спирты.
Н2
Спрос на продукты оксосинтеза линейного строения, использующиеся для получения поверхностно активных веществ, пластификаторов, растворителей, гораздо больше, чем на соответствующие разветвленные альдегиды и спирты. В связи с этим было важно найти условия, обеспечивающие получение с высокой селективностью продуктов линейного строения. Впервые использованные Роеленом в 1938 г. кобальтовые катализаторы (Со2(СО)8 и его производные) в оптимальных условиях (120-160о, 20-35 МПа, СО: Н2=1) при гидро-формилировании пропилена обеспечивали соотношение линейных и разветвленных продуктов (н: изо), равное примерно четырем. Этого было недостаточно. Понижение температуры приводило к некоторому увеличению селективности образования линейных продуктов, но уменьшало скорость их образования. Увеличение давления синтез-газа свыше 350 атм незначительно повышало соотношение н: изо, но существенно увеличивало энергетические затраты и материалоемкость производства.
Кинетическая модель процесса соответствует механизму с участием гидридных комп-
R = k[алкен][Co]PH2/PCO
лексов кобальта, образующихся при взаимодействии металлического кобальта и(или) октакарбонила кобальта с синтез-газом. Cчитают, что уменьшение скорости процесса с увели-
Н2
2Со + 8СО ↔ Со2СО8 ↔ 2НСо(СО)4
НСо(СО)4 ↔ НСо(СО)3 + CO
НСо(СО)3 + RCH=CH2 ↔ HCo(CO)3(RCH=CH2) - π -комплекс
HCo(CO)3(RCH=CH2) à RCH2CH2Co(CO)3 – σ 1 – комплекс
HCo(CO)3(RCH=CH2) à RCH(Co(CO)3)CH3 – σ 2 – комплекс
σ 1 + CO + Н2 à RCH2CH2CHO + НСо(СО)3
σ 2 + CO + Н2 à RCH(CHO)CH3 + НСо(СО)3
чением парциального давления оксида углерода связано, в основном, со стадией выделения одной молекулы СО из тетракарбонилгидрида кобальта с освобождением координационного места для алкена. Соотношение н: изо определяется соотношением скоростей внедрения алкена по правилу Марковникова и против него. Поскольку гидридный комплекс кобальта может рассматриваться в качестве кислоты, то его присоединение к двойной связи α -алкена по правилу Марковникова (водород – к наиболее гидрированному атому углерода двойной связи) приведет к разветвленному продукту, а против этого правила – к образованию линейного продукта гидроформилирования. Основным фактором, определяющим региоселективность процесса, являются, по-видимому, стерические препятствия на стадии внедрения алкена и соотношение скоростей внедрения оксида углерода в связь Со-С. Стерические препятствия, возникающие из-за объемных лигандов, координированных кобальтом, стимулируют присоединение кобальта (с лигандами) к концевому атому углерода алкена. Действительно, увеличение давления СО приводит к большему насыщению координационной сферы кобальта, увеличению стерических препятствий и соотношения н: изо в продуктах. К еще большему увеличению селективности приводит введение более объемных, чем оксид углерода, фосфиновых лигандов (см. табл. 11). Но к наибольшему увеличению селективности привело использование родиевых катализаторов, модифицированных арилфосфиновыми лигандами (табл. 11).
В качестве катализаторов в промышленности используют соединения кобальта и родия. Условия и показатели процесса оксосинтеза существенно зависят от природы используемого катализатора (табл.11). На основе этих данных в начале 80-х считалось, что при гидроформилировании низших алкенов технологические схемы классического оксосинтеза с использованием октакарбонила кобальта, как минимум, конкурентоспособны по сравнению со схемами, использующими родиевые катализаторы. Однако последующее развитие событий опровергло этот вывод. Интересна динамика изменения объема производства продуктов оксосинтеза и доли процессов, использующих родиевые катализаторы (табл.12).
Таблица 11
Показатели процесса гидроформилирования пропилена при использовании разных катализаторов
| Показатели | Нафтенатно-испарительные схемы | Процесс с кобальтфосфиновым катализатором | Процесс с родийфосфино-вым катализатором | |
| ВНИИНЕФ-ТЕХИМ-Leuna Werke | C рециклом алкена (ВНИИНЕФТЕХИМ) | |||
| Катализатор | НСо(СО)4 | НСо(СО)4 | НСо(СО)3РBu3 | HRh(CO)(PPh3)3 |
| Температура, оС | 120-160 | 120-130 | 160-200 | 60-120 |
| Концентрация катализатора, %масс. | 0, 1-0, 5 | 0, 1-0, 3 | 0, 6 | 0, 01 |
| Степень гидрирования алкена | Низкая | Низкая | Высокая | Высокая |
| Продукты реакции | Альдегиды и спирты | Альдегиды | Спирты | Альдегиды |
| Отношение продуктов, н: изо | 80: 20 | 80: 20 | 88: 12 | 92: 8 |
| Селективность (%) по линейным продуктам сумме цел. продуктов |
Таблица 12
Динамика изменения объема производства продуктов оксосинтеза и используемости катализаторов различного типа
| Год | Объем производства, млн.т/г | В том числе на родиевых катализаторах, % |
| 0, 8 | ||
| 5, 2 | ||
| 6, 6 |
Приведенные данные свидетельствуют о существенной переориентации даже действующих производств на родиевые катализаторы.
В процессах оксосинтеза возникают и требуют решения наиболее важные проблемы гомогенного металлокомплексного катализа: проблемы выбора каталитической системы и оптимизации условий синтеза для получения максимальной селективности и приемлемой производительности, сложная ситуация с разделением достаточно высококипящих продуктов и каталитической системы, обладающей в условиях синтеза определенной летучестью, задача поиска эффективной методики регенерации катализатора. На примере этого процесса были проверены различные варианты решения указанных задач. Поэтому он, наряду с другими, использован в качестве иллюстрации вышеописанных подходов к организации технологии гомогенно-каталитического процесса. Технология процесса не будет рассматриваться в целом (более подробно технология этого процесса рассмотрена в курсе «Технология основного органического синтеза»), основное внимание будет уделено организации реакционного узла, решению проблемы разделения каталитической системы и продуктов реакции и регенерации катализатора.
Процесс оксосинтеза протекает в гетерофазной системе газ-жидкость. Конструкция реактора должна в связи с этим обеспечивать достаточную скорость растворения газообразных реагентов (оксида углерода, водорода и алкена при гидроформилировании низших алкенов) в жидкой фазе. Обычно это обеспечивается использованием различных барботажных устройств. В качестве основного реактора в процессе оксосинтеза используют барботажную колонну. Этот простой по устройству аппарат обеспечивает достаточный контакт газовой и жидкой фаз. Недостатки колонных аппаратов связаны с неоптимальной функцией распределения времени пребывания жидкой фазы, и, как следствие, уменьшением селективности процесса за счёт протекания побочных реакций превращения продуктов гидроформилирования.
Процесс гидроформилирования сильно экзотермичен. Например, экзотермический тепловой эффект синтеза н-масляного альдегида из пропилена составляет примерно 117 кДж/моль. Повышение температуры выше оптимальной нежелательно, так как при этом уменьшается селективность по линейным продуктам. Необходимую температуру поддерживают за счет введения холодного сырья или использования внешнего теплообмена (устройства типа трубок Фильда или встроенные трубчатки).
Предложены более сложные реакторы гидроформилирования, включающие две секции. Первая секция близка к реактору полного смешения и обеспечивает быстрое карбонилообразование и уменьшение индукционного периода, вторая секция по типу ближе к реактору вытеснения и обеспечивает максимальную степень превращения реагентов при минимальном времени пребывания. Последнее важно, поскольку альдегиды склонны к последовательным побочным реакциям.
4.3.1 Схемы с термической декобальтизацией (выделение кобальта в виде металла на поверхности твердой фазы носителя)
Рассмотрим технологические схемы оксосинтеза в историческом аспекте. Исторически первой была предложена группа технологических схем, основанных на термической декобальтизации, т.е. выделение кобальта из контактного раствора проводили за счет перевода его в металлическое состояние (твердую фазу) с адсорбцией на сорбенте со смещением равновесия реакции карбонилообразования влево. Смещение равновесия обеспечивается уменьшением парциального давления монооксида углерода и повышением
H2
2Со + 8СО ↔ Со2(СО) 8 ↔ 2НСо(СО)4
температуры.
Первой была предложена так называемая диадная схема, включающая две одинаковые колонны высокого давления 1, 3, заполненные насадкой (рис. 10).
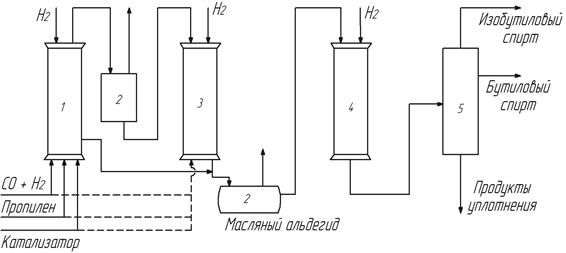
Рис. 10. Диадная схема оксосинтеза
В одной из колонн перед началом процесса на насадке находится нанесенный кобальт(0). Оба аппарата снабжены всеми коммуникациями: подводом синтез-газа, алкена, водорода и связаны между собой через фазовый сепаратор 2. Каждая из колонн связана со сборником-накопителем 2 жидких продуктов гидроформилирования (рис. 10). В реактор 1, заполненный насадкой с нанесенным кобальтом, подают синтез-газ под необходимым давлением и алкен. Происходит растворение кобальта с поверхности насадки с образованием карбонилов. Вышеописанное равновесие смещается вправо, начинается процесс гидроформилирования. Контактный раствор с определенной концентрацией продуктов через фазовый сепаратор 2, в котором отделяют непрореагировавший синтез-газ, отводят в аппарат 3 с чистой насадкой. В аппарате 3, выполняющем в этот период функцию декобальтизера, при температуре примерно 300о и давлении водорода 2, 5 МПа карбонильные комплексы разрушаются, равновесие карбонилообразования смещается влево, и кобальт(0) садится на поверхность насадки. Освобожденный от кобальта жидкий продукт поступает в сборник-накопитель 2. Цикл продолжается до тех пор пока большая часть кобальта не окажется на насадке в аппарате 3. После этого функции аппаратов 1 и 3 меняются. Третий аппарат становится реактором карбонилообразования и гидроформилирования, а первый – декобальтизером. В ходе термической декобальтизации в атмосфере водорода происходит частичное гидрирование альдегидов в спирты. В связи с этим в качестве целевых продуктов в этом случае являются спирты. Органические продукты из приемника 2 поступают в реактор гидрирования 4, в котором остаточные количества альдегидов превращаются в спирты, и далее в систему разделения.
Недостатки диадной схемы (неэффективность использования реакторов высокого давления, периодичность в получении продуктов оксосинтеза) были частично устранены в триадной схеме (рис. 11). По этой схеме получение карбонилов кобальта и гидроформилирование проводят в разных аппаратах. Используют три аппарата высокого давления: кобальтизер, в котором образуется раствор карбонилов кобальта в инертном (лучше углеводородном) растворителе 3’ или 3”, собственно реактор гидроформилирования 1 и декобальтизер 3’ или 3”, где карбонилы кобальта разрушаются и металлический кобальт извлекается из контактного раствора. Так же, как в диадной схеме, кобальтизер заполняется насадкой (например, пемзой) с нанесенным на неё кобальтом, а декобальтизер - чистой насадкой. По мере истощения кобальта в кобальтизере и накопления его в декобальтизере функции этих аппаратов меняются. Все коммуникации в аппаратах 3’ и 3” дублируются. Общими недостатками диадной и триадной схем являются цикличность работы, т.е. необходимость периодического переключения потоков (не реже, чем один раз в сутки) и нерациональное использование аппаратов высокого давления.
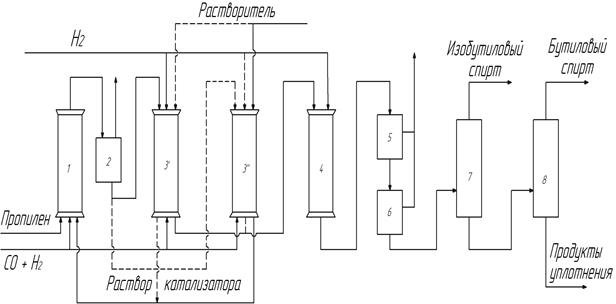
Рис. 11. Триадная схема оксосинтеза
Принцип термического разложения карбонилов кобальта (вывод кобальта в твердую фазу) был использован в схеме с осаждением кобальта на движущийся носитель с развитой поверхностью (кизельгурная схема, рис. 12). Кобальт вводится в процесс в виде суспензии порошка носителя с нанесенным металлическим кобальтом. Схему назвали кизельгурной по названию использовавшегося природного алюмосиликатного носителя.
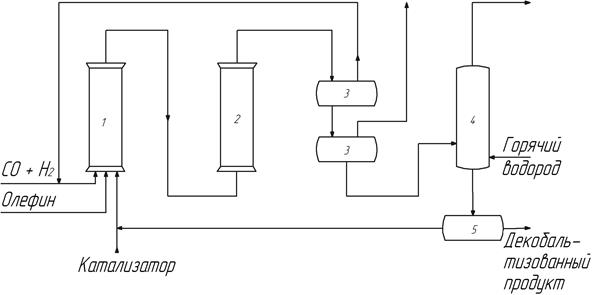
Рис. 12. Кизельгурная схема оксосинтеза
Процесс проводят в двух последовательно соединенных реакторах 1 и 2 (рис. 12). В первом реакторе при 25-30 МПа синтез-газа и 150-170о происходит образование карбонилов кобальта и частично процесс гидроформилирования. Заканчивается процесс гидроформилирования во втором реакторе. Для интенсификации синтеза температура во втором реакторе поддерживается на 10-20 градусов выше, чем в первом. Жидкая фаза (после отделения от синтез-газа в фазовых сепараторах 3) с суспендированным носителем поступает на стадию декобальтизации, которую проводят в двух последовательно соединенных реакторах 4 при 120-130 о и давлении водорода 2, 5-3 МПа. Кобальт в этих условиях осаждается на носитель и отделяется от жидкой фазы с помощью магнитных сепараторов 5.
Достоинствами этой схемы являются полная непрерывность и эффективное использование аппаратов высокого давления. Недостатки – повышенная эррозия аппаратуры и запорной арматуры из-за абразивных свойств суспензии и сложность отделения твердых частиц от жидкой фазы с помощью магнитных сепараторов.
Особенностью всех технологических схем с термической декобальтизацией является частичное гидрирование альдегидов в спирты на стадии декобальтизации из-за контакта с металлическим кобальтом (хорошим катализатором гидрирования) под давлением водорода. Поэтому эти схемы целесообразно использовать для производства спиртов.
4.3.2 Солевые схемы (выделение металла в виде соли за счет экстракции)
Схемы декобальтизации, основанные на экстракции, называют солевыми. Лучшим экстрагентом для извлечения солей кобальта (см. вышеприведенные требования) оказалась вода. Для экстракции соединений кобальта водой и уменьшения его растворимости в органическом растворе нужно перевести карбонилы кобальта в соль. Это достигается действием окислителя в присутствии кислоты. В качестве окислителей используют кислород воздуха, пероксид водорода, азотную кислоту. В качестве кислот - муравьиную, уксусную и азотную кислоту. Образующаяся соль кобальта(II) переходит в водную фазу и в виде водного раствора подается на стадию карбонилообразования.
Со2(СО)8 + О2 + 4НХ à 2CоХ2 + 8СО + 2Н2О
3Со2(СО)8 + 16НNО3 à 6Cо(NО3)2 + 4NО + 8Н2О + 24СО
На стадии карбонилообразования происходит взаимодействие соли кобальта(II) c синтез-газом с восстановлением кобальта в карбонильные соединения.
2СоХ2 + 8СО + 2Н2 à Со2(СО)8 + 4НХ
2СоХ2 + 8СО + 3Н2 à 2НСо(СО)4 + 4НХ
Если воду на стадии декобальтизации не добавляют, то соль кобальта может образовать суспензию и отделяться от органических продуктов фильтрованием или центрифугированием. В этом случае соль кобальта возвращают на стадию карбонилообразования в виде суспензии в инертном растворителе, используемом в процессе (рис. 13).
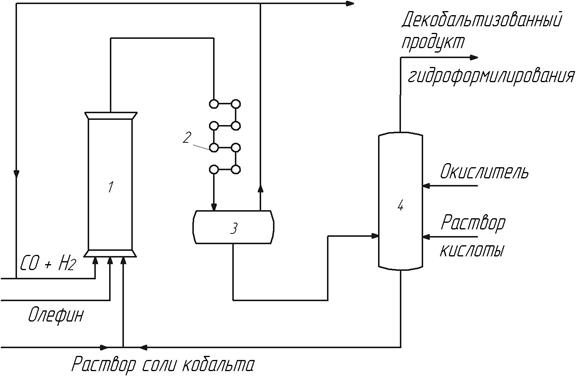
Рис. 13. Солевая схема оксосинтеза
К солевым схемам можно отнести и так называемую кульмановскую схему, в которой не используется окислитель, а главную роль играют кислотные свойства гидрокарбонила кобальта (рис. 14). Процесс протекает в реакторе 1 в обычных для этого катализатора условиях: 110-180о и давлении синтез-газа 20-25 МПа. После необходимого времени пребывания в реакторе (1 – 2 ч) контактный раствор попадает в колонну 2, в которой контактирует с водным раствором соды. При этом происходит реакция нейтрализации гидрокарбонила кобальта и экстракция образовавшегося солеобразного карбонила в водную фазу.
НСо(СО)4 + Nа2СО3 à NаСо(СО)4 + NaHСО3
Продукты из колонны 2 поступают в фазовый сепаратор 3, в котором удаляются остаточные газы и разделяются органическая и водная фазы. Органическая фаза поступает в следующую экстракционную колонну 2, где отмывается водой от остатков катализатора и следует в систему разделения (на рис. не показана). Водные фазы смешиваются и поступают в колонну 4, в которой подкисляются серной кислотой. Снизу в колонну 4 подают синтез-газ. В колонне 4 регенерируется летучий и умеренно растворимый в кислоте гидрокарбонил кобаль-
2 NаСо(СО)4 + Н2SО4 à 2 НСо(СО)4 + Nа2SО4
та и в токе синтез-газа возгоняется в колонну 5. Туда же подают алкен и растворитель, в растворе которых гидрокарбонил кобальта поступает в реактор 1 для проведения гидроформилирования (рис. 14).
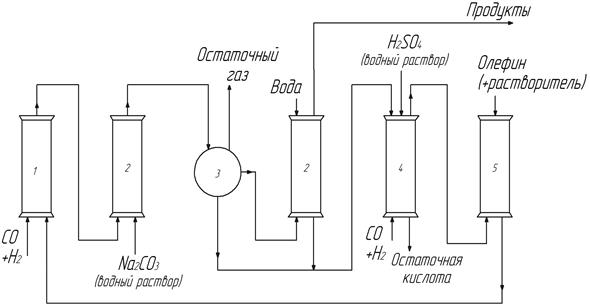
Рис. 14. Кульмановская схема оксосинтеза
Достоинством солевых схем является отсутствие необходимости использовать реактор высокого давления на стадии декобальтизации, а также сохранение альдегидов в качестве основных продуктов оксосинтеза. Более эффективны эти схемы в применении к гидроформилированию высших алкенов в альдегиды. Недостатки этих схем связаны с применением дополнительных реагентов, появлением сточных вод и усилением коррозии оборудования.
4.3.3 Испарительные схемы (отделение продуктов за счет отгонки)
В начальный период разработки технологии оксосинтеза для процессов на основе немодифицированных кобальтовых катализаторов предпринимались попытки разработать стадию декобальтизации, основанную на разной летучести карбонильных соединений кобальта и альдегидов – продуктов оксосинтеза. Для этого предполагалось сместить равновесие между октакарбонилом и гидрокарбонилом кобальта в пользу менее летучего октакарбонила за счет понижения давления водорода и повышения давления оксида углерода (для предотвращения распада карбонильных соединений до металлического кобальта). Пред-
Со2(СО)8 + Н2 ↔ 2НСо(СО) 4
полагалось, что при этом разница в летучести соединений кобальта и альдегидов окажется достаточной для проведения отгонки продуктов в токе оксида углерода. Однако, такая схема оказалась не эффективна и не нашла промышленного применения. Гораздо более перспективным оказался вариант, при котором отделению продуктов ректификацией предшествовал перевод кобальта в нелетучее и каталитически неактивное состояние. Это состояние достигалось окислением кобальта в соли органических (например, нафтеновых) кислот для обеспечения их растворимости в органической фазе. Принцип работы нафтенатно-испарительной схемы понятен из рис. 15.
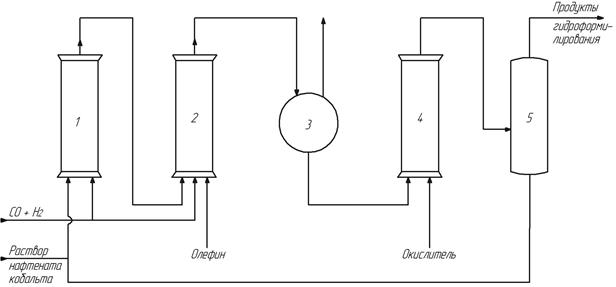
Рис. 15. Нафтенатно-испарительная схема оксосинтеза
Реакции карбонилообразования (реактор 1) и окисления кобальта (реактор 4) полностью аналогичны реакциям, приведенным выше для солевой схемы. В ректификационной колонне 5 продукты оксосинтеза отгоняют от стабилизированного раствора нафтенатов кобальта в высококипящем растворителе. Кубовый остаток из колонны 5 возвращают в реактор 1. Потери кобальта не превышают одного процента.
Отделять ректификацией продукты гидроформилирования от контактного раствора без его дополнительной обработки можно в случае сравнительно малолетучих каталитических систем, на основе кобальта и родия, включающих фосфорсодержащие лиганды (см. табл. 11). При этом отгоняемые продукты должны быть низкокипящими, содержащими не более четырех атомов углерода. Например, фирма «Юнион Карбайд Компани» изучила возможность реализации двух схем этого типа. Первой для родиевого катализатора была проверена возможность организации совмещенного процесса, т.е. возможности отогнать продукты гидроформилирования из реактора в условиях процесса за счет увеличения скорости подачи исходных реагентов (рис. 16).
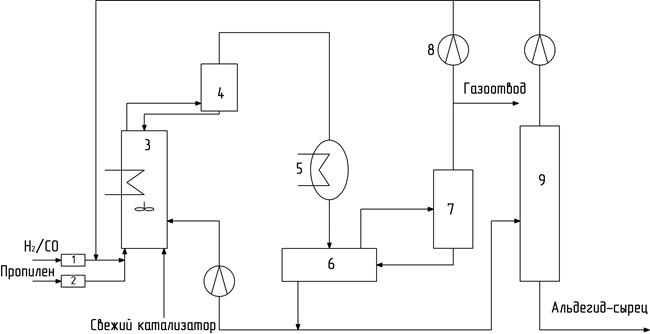
Рис. 16. Схема фирмы Юнион Карбайд Компани с рециклом газа
Синтез-газ и пропилен проходят через системы очистки 1 и 2, соответственно, и поступают в реактор 3, в который непрерывно подают регенерированный катализатор. Потери катализатора восполняются за счет свежего каталитического раствора. Непрореагировавший газ вместе с продуктами гидроформилирования выводят из реактора через каплеотбойник 4 и холодильник-конденсатор 5 в фазовый сепаратор 6. В последнем происходит разделение жидкой и газовой фаз. Газовая фаза поступает в холодильник-конденсатор 7, откуда несконденсировавшаяся часть возвращается в реактор. Конденсат направляют в сепаратор 6. Жидкую фазу из сепаратора 6 разделяют на два потока. Первый возвращают в реактор, а второй направляют в ректификационную колонну 9. Из куба колонны 9 отводят продукты гидроформилирования в систему разделения, пар из верха колонны возвращают в реактор. Определенная часть рециркулирующего газа отводится из цикла для предотвращения накопления инертных примесей. Недостатком этой схемы является большой расход энергии на рецикл газа.
Второй вариант, проверенный той же фирмой, включает рецикл жидкой фазы (рис. 17). Жидкая фаза из реактора 1 отводится в фазовый сепаратор 2. Газовую фазу возвращают в реактор. Жидкую фазу дросселируют через вентиль 3 в колонну 4, в которой при 25о и давлении 0, 1-0, 2 МПа выделяется большая часть растворенного синтез-газа и пропилена, рециркулируемых затем в реактор 1. Жидкая фаза из колонны 4 поступает в колонну 5, где под вакуумом (0, 015 МПа) и при нагревании (130о) происходит испарение остатков синтез-газа, пропилена и основной части альдегидов (продуктов оксосинтеза).
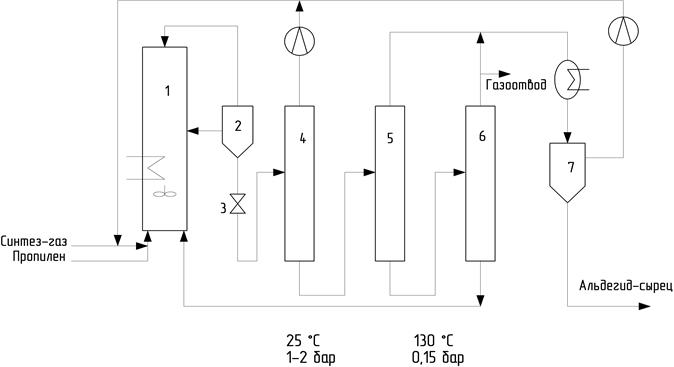
Рис. 17. Схема оксосинтеза фирмы Юнион Карбайд Компани с рециклом жидкой фазы
Эта схема, судя по имеющимся данным, оказалась более экономична, чем предыдущая.
4.3.4 Оксосинтез в двухфазных системах вода-органический растворитель
Гидроформилирование алкенов можно проводить в системах, содержащих воду и несмешивающийся с ней органический растворитель, служащий экстрагентом для продуктов процесса. В таких случаях обычно используют каталитические системы, включающие комплексы родия с водорастворимыми фосфиновыми лигандами. Хорошая растворимость комплексов родия в воде и нерастворимость в органическом растворителе достигается за счёт использования сульфированных фосфинов. Гомогенно-каталитический процесс протекает в водной фазе, а продукты экстрагируются в органическую фазу. В качестве промышленного процесса такого типа можно привести вариант технологии, разработанный фирмами Рурхеми и Рон Пуленк (Ruhrchemie - Rȏ ne Poulenc), в котором применяют в качестве лиганда натриевую соль трифенилфосфин-три-метасульфоната P(PhSO3Na)3 (TPPTS). Растворимость TPPTS в воде составляет 1200 г/л.
Упрощенная технологическая схема процесса Рурхеми – Рон Пуленк, используемого для получения масляных альдегидов, включает реактор (1), фазовый сепаратор для разделения жидких фаз (2), холодильник (3), подсистему разделения (4, 5) (рис. 18). Водная фаза, содержащая комплекс родия, из сепаратора (2) через холодильник (3) возвращается в реактор (1). Органический слой, содержащий н-масляный и изомасляный альдегиды, отделяют в сепараторе и подают на разделение.
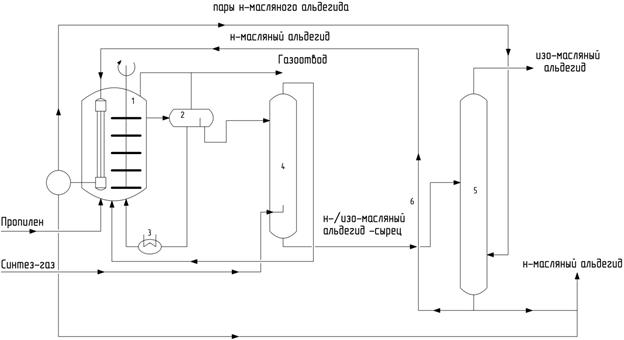
Рис. 18. Схема оксо-процесса в двухфазной системе фирм Рурхеми и Рон-Пуленк
В промышленности процесс гидроформилирования с использованием водорастворимого катализатора Rh-TPPTS применяется c 1984 г. до настоящего времени фирмой Рурхеми для производства бутиловых и 2-этилгексилового спиртов с суммарной мощностью 620000 т/г, а также амиловых и 2-пропилгексиловых спиртов (12000 т/г).
К преимуществам процесса с использованием катализатора Rh-TPPTS по сравнению с системой Rh-PPh3 относятся простота технологии разделения катализатора и продуктов (потери катализатора с продуктами менее 1 ppm), низкое соотношение лиганд: родий (~10), обеспечивающее более высокую региоселективность. К недостаткам этого процесса относят высокие скорости потока раствора катализатора через реактор, повышенные концентрации родия и несколько более жесткие условия реакции. Кроме того, применимость этого варианта технологии имеет ограничения по сырью, а именно, низкая растворимость в воде алкенов с длиной углеродной цепи более 4 не позволяет достигать практически приемлемых скоростей гидроформилирования.