Главная страница Случайная страница
КАТЕГОРИИ:
АвтомобилиАстрономияБиологияГеографияДом и садДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеталлургияМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРелигияРиторикаСоциологияСпортСтроительствоТехнологияТуризмФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Гіпертензивна хвороба. 1 страница
|
|
Гіпертензивна хвороба (ГХ) одна із форм артеріальної гіпертензії (АГ) є найбільш поширеним захворюванням. В Україні АГ страждають майже 13 мільйонів людей. Серед осіб з підвищеним артеріальним тиском (АТ) знають про наявність АГ 62% хворих, з них лікуються лише 23, 3%, причому ефективно лише 12, 8%. У осіб з високим АТ у 3-4 рази частіше розвивається ІХС і в 7 разів частіше - порушення мозкового кровоплину. Економічні збитки, зумовлені тимчасовою непрацездатністю, інвалідністю та передчасною смертністю від АГ, та зв’язаних з нею церебро-васкулярних захворювань перевищують 2 мільяарди гривен в рік.
Таким чином, проблема АГ є сьогодні національною проблемою. Указом Президента України в 1999 році введена Національна Програма профілактики і лікування АГ в Україні, метою якої є зниження захворюваності населення на АГ, ІХС, ЦВЗ, смертності від ускладнень АГ, підвищення тривалості і якості життя хворих на серцево-судинні захворювання.
Досягнення цієї мети передбачає вирішення наступних завдань: пропаганда здорового стилю (способу) життя; зміцнення первинної ланки охорони здоров’я кадрами і ресурсами, необхідними для здійснення медико-санітарної освіти населення, виявлення хворих на АГ, профілактики АГ та її ускладнень; створення стандартів діагностики і лікування на підставі наукових даних, забезпечення ефективної діагностичної, лікувальної, реабілітаційної допомоги хворим на АГ та її ускладнення; забезпечення населення тонометрами і ефективними антигіпертензивними ліками.
Основні фізіологічні системи контролю артеріального тиску. Серед механізмів, які забезпечують формування артеріального тиску (АТ) і підтримку його на нормальному рівні умовно виділяють: гемодинамічні фактори, які визначають висоту АТ, і нейрогуморальні системи, які впливають на гемодинамічні фактори в такому напрямку, щоб утримати АТ у фізіологічних межах.
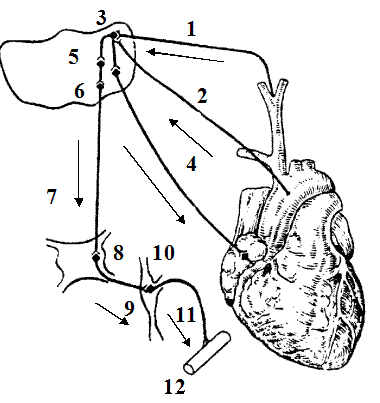 |
Рис. 10. Схема барорецепторної рефлекторної дуги
(адаптовано за М.С.Кушаковським, 1995)
1 - ramus caroticus; 2 - аферентні волокна n.vagi, що виходять з дуги аорти;
3 - nucleus tractus solitary (NTS); 4 - еферентні волокна n.vagi, що йдуть до серця;
5 - центральні альфа- адренорецептори в зоні NTS; 6 - інгібіторні нейрони;
7 - бульбоспінальні шляхи; 8 - nucl. іntermediolateralis в бокових рогах спинного мозку; 9 - прегангліонарні волокна; 10 - межовой стовбур;
11 - постгангліонарні волокна; 12 - кровоносні судини.
До гемодинамічних факторів, які визначають рівень АТ відносяться: 1) серцевий викид або хвилинний об’єм (ХО) серця, тобто кількість крові, яка поступає в судинну систему за хвилину роботи серця; 2) загальний периферичний судинний опір (ЗПСО) або прохідність резистивних судин (артеріол і прекапілярів), від якої залежить інтенсивність відтоку крові з артеріального русла; 3) напруженість стінок аорти та її великих гілок, яка створює загальний еластичний опір; 4) в’язкість крові.
Основний вплив на рівень АТ мають ХО і ЗПО, оскільки у здорових людей в’язкість крові є практично величиною постійною. Що ж торкається загального еластичного опору, то він підвищується головним чином у осіб похилого і старечого віку, коли ущільнюються стінки аорти і великих артерій еластичного типу.
Між ХО і ЗПСО існують реципрокні взаємовідносини: при збільшенні ХО у здорової людини адекватно понижується ЗПСО (настільки, що вплив збільшеного ХО на АТ урівноважується). Рівень ХО залежить від об’єму циркулюючої плазми (ОЦП), який у свою чергу тісно пов’язаний з об’ємом інтерстиціальної рідини. Ці взаємовідносини регулюються складною нейро-гуморальною системою і насамперед симпатичними нервами.
Стан ЗПСО в найбільшій мірі визначається інтенсивністю вазоконстрикції. У свою чергу величина просвіту резистивних судин контролюється складними нейро-гуморальними та ендокринними механізмами, суть яких коротко зводиться до наступного.
Всі регуляторні біокібернетичні механізми регуляції АТ зосереджені у двох основних системах: 1) короткочасної дії або адаптаційній контрольній системі; 2) тривалої дії або інтегральній контрольній системі.
Система короткочасної дії. Представлена декількома регуляторними механізмами зворотного зв’язку, найважливішими з яких є: а) барорецепторний рефлекс – “барорецептори великих артерій” ® центри головного мозку (nucleus tractus solitarii, Caudal Medulla, Rostral Medulla) ® симпатичні нерви ® резистивні і ємкісні судини, серце ® АТ; б) нирковий механізм (юкстагломерулярний апарат ® ренін ® ангіотензин ІI ® резистивні судини ® АТ).
В нормі 80% реніна поступає в кров у неактивній формі (проренін). Точно не відомий фермент, який сприяє перетворенню прореніна у ренін (можливо це здійснює плазмовий калікреїн). Пошкоджена ж нирка секретує активний ренін (бета-адреноблокатори гальмують секрецію активного реніна, але не впливають на виділення прореніна). Регуляція секреції реніна здійснюється різними шляхами, зокрема через: 1) барорецептори, що є у стінці привідних ниркових артеріол, які чутливі до зміни ниркового перфузійного тиску (секреція реніна зростає, якщо цей тиск зменшується); 2) хеморецептори “каменистого тільця” (macula densa) дистальних ниркових канальців, які реагують на коливання концентрації NaCl в рідині, що поступає в канальці (секреція реніна зростає при зниженні транспорта іонів Cl- і гальмується при їх надлишку); 3) ниркові симпатичні нерви, які закінчуються в мембрані юкстагломерулярних клітин, на яких розташовані b1-адренорецептори (посилення b-адренорецепторних адренергічних сигналів супроводжується збільшенням секреції реніна).
Разом з тим, на утворення і виділення реніна мають вплив і ниркові простагландини (Pg) - особливо стимулює секрецію реніна PgI2 (простациклін), який синтезується корою нирок. Ефект PgI2 опосередковується через барорецептори юкстагломерулярних клітин. Секреція реніна залежить також від концентрації в крові катехоламінів, глюкагона, іонів Na+ і К+, нарешті, ангіотензина ІІ (А-ІІ).
Початком утворення АІІ є взаємодія реніна з плазмовим білком, який виробляється печінкою. Його називають субстратом реніна або ангіотензиногеном, з якого й утворюється ангіотензин І (АІ). Вироблення ангіотензиногена зростає при вагітності, цирозах печінки, надлишку естрогенів, прийомі глюкокортикоідів або тироксину, а також при деяких формах гострого і хронічного запалення.
АІІ виникає внаслідок впливу на АІ ангіотензинперетворюючого фермента (АПФ), який розташований на мембранах клітин мікросудин. Однак, тепер з’ясувалось, що АІ може перетворюватись в АІІ не лише під впливом АПФ, але й хімази, катепсину G, передсердного натрійуретичного фактора. Це так званий не-АПФ-залежний механізм утворення АІІ. При цьому йдеться про те, що АІІ може утворюватись безпосередньо з ангіотензиногену. Активують утворення АІІ з ангіотензиногена: тонін слинних залоз, катепсин G, тканинний активатор плазміногена (ТАП). Цим, очевидно й пояснюється неефективність у деяких хворих інгібіторів АПФ.
Ефекти АІІ здійснюються через відповідні клітинні рецептори. Розрізняють декілька типів рецепторів до АІІ, зокрема, АТ1А, АТ1В, АТ2, АТ3, АТ4. Основні ефекти АІІ здійснюються через рецептори типу АТ1А, АТ1В. Судинозвужуюча активність АІІ в 50 разів вища, ніж у норадреналіна.
Між рівнем АІІ в плазмі і активністю юкстагломерулярних клітин існують реципрокні взаємовідносини. Зменшення АІІ в плазмі крові компенсується посиленням секреції реніна з подальшим утворенням АІІ і, навпаки, при надлишку А ІІ в плазмі секреція реніна юкстагломерулярними клітинами гальмується.
АІІ розщеплюється з участю плазмових ферментів - ангіотензиназ з утворенням неактивних олігопептидів, а також ангіотензина ІІІ (АІІІ).
Система тривалої дії (інтегральна). Ця система регуляції АТ складається з численних механізмів, серед яких провідне значення має: механізм “нирки-кора надниркових залоз (альдостерон) - концентрація іонів Nа+ – рідке середовище організма”. Всі елементи ренін-ангіотензин-альдостеронової системи (РААС) знайдені не лише у гемоциркуляції, але і в багатьох тканинах і органах, перш за все у головному мозку та серці. Синтез ангіотензиногена, реніна, ангіотензинів відбувається в цих органах внутрішньоклітинно. Локальні механізми здатні тривало впливати на резистивні судини, регулюючи їх прохідність, а відповідно - ЗПСО і АТ. Локальні системи “ренін-АІІ” мало впливають на рівні реніна і АІІ, які циркулюють у крові.
Вплив АІІ на баланс іонів Nа+ і рідке середовище - найважливіша функція інтегральної, тривалодіючої системи регуляції АТ. Посилення реабсорбції Nа+ забезпечується: а) безпосереднім впливом АІІ на ниркові канальці; б) опосередкованим посиленням секреції альдостерона клітинами клубочкового пласту кори наднирників. Швидкість біосинтезу і виділення альдостерона регулюється АІІ і АІІІ, АКТГ, іонами К+ і Nа+ в плазмі крові. Навіть невелике підвищення концентрації К+ може на тривалий час посилити секрецію альдостерона. Цей гормон в основному й забезпечує гомеостаз іонів Nа+, К+ і води.
Депресорні механізми інтегральної системи регуляції артеріального тиску. З’ясувалось, що нирки володіють не лише пресорними, але і депресорними властивостями. Основними компонентами депресорної системи нирок є системи простагландинів і калікреїнів.
Простагландини - це ненасичені циклічні жирні кислоти, продукти метаболізму арахідонової кислоти, вони протидіють гормональній ангіотензин-II-зумовленій і α -адрененергічній (норадреналіновій) вазоконстрикції. Вони затримують виділення норадреналіна із закінчень симпатичних нервів, регулюють вміст циклічних нуклеотидів (цАМФ, цГМФ), взаємодіють з кальцієвим механізмом гладком’язових клітин судинних стінок. Синтезуються ниркові простагландини, в основному, в мозковому пласті нирок. Найбільше значення мають ниркові РgІ2 (простациклін), PgG2, РgЕ2.
Калікреїн-кінінова система нирок. Калікреїн синтезується епітелієм дистальних канальців нирок. Внаслідок взаємодії з кініногенами утворюється лізил-брадикінін (калідин), який і складає головну масу ниркових кінінів. Лізил-брадикінін, як і брадикінін плазми, руйнується кініназами І і ІІ (остання ідентична ангіотенгинперетворюючому ферменту, який перетворює АІ в АІІ). Кініни посилюють нирковий кровоплин, натрій- і гідроурез, що особливо треба пам’ятати при застосуванні інгібіторів АПФ (капотен, еналаприл, фозіноприл тощо). Усі три гормональні системи нирок - ренін-ангіотензинова (пресорна), простагландинова і калікреїн-кінінова (депресорні) - взаємозв’язані. Виділення реніна і простагландинів в нирках зростає у відповідь на зниження ниркового перфузійного тиску крові. Блокада синтеза РgІ2 індометацином обмежує утворення реніна.
Судинні депресорні механізми. Ендотеліальні клітини судин здатні секретувати не лише судинозвужуючі (ендотеліни), але й судинорозширювальні речовини, зокрема, ендотелій-релаксуючий фактор. Він ідентифікований як оксид азоту (NO). Синтез NO з амінокислоти L-аргініну здійснюється в присутності іонів Са++ і кальмодуліна в якості кофактора. NO активує цитоплазматичну (розчинну) форму гуанілатциклази і в такий спосіб сприяє посиленому утворенню вазодилататорного цГМФ. Крім розслаблення гладком’язових клітин судинної стінки NO гальмує агрегацію тромбоцитів.
При есенціальній гіпертензії пошкоджені і інші вазодилататорні механізми. Насамперед це передсердний натрійуретичний фактор (ПНУФ) серця і мозку. Він гальмує активацію симпатичної нервової системи, утворення реніна в нирках, секрецію альдостерона і вазопресина, затримку іонів Na+ і води. ПНУФ обмежує гіпертрофію міокарда і гладком’язових клітин резистивних судин. Його нестача сприяє прогресуванню діастолічної гіпертензії шляхом підвищення ЗПСО. У хворих на есенціальну гіпертензію навіть незначне зростання концентрації ПНУФ в плазмі призводить до посилення натрійурезу. ПНУФ є важливою складовою багатогранної депресорної системи.
У плазмі крові знаходиться ще один депресорний чинник, – дигіталісоподібний натрійуретичний фактор, – який приймає участь в регуляції реабсорбції Nа+ в ниркових канальцях і впливає на прохідність резистивних судин.
З інших депресорних механізмів має значення в регуляції АТ допамінергічний депресорний механізм. Допамін ідентифікований як попередник норадреналіна. Однак тепер з’ясовано, що він має самостійне значення як нейромедіатор ЦНС і на периферії. Фізіологічна роль допаміна полягає в контролі за екскрецією натрія з сечею, тому є важливим компонентом депресорної системи нирок. Зниження вмісту допміна в нирках гальмує виділення натрія.
Таким чином, організм людини володіє комплексом пресорних і депресорних механізмів як ниркового, так і позаниркового походження, добре збалансованих у фізіологічних умовах.
Сучасні уявлення про етіологію і патогенез гіпертензивної (гіпертонічної) хвороби. Найбільш поширеною в етіопатогенезі ГХ є концепція центрального норадреналіндефіцитного шляху розвитку цього захворювання, у витоках якої лежить центрогенно-нервова теорія Г.Ф.Ланга (1935). З точки зору Г.Ф.Ланга та О.Л. Мяснікова ГХ є наслідком психічного перевантаження людини, впливу на його психічну сферу емоцій від’ємного характера (психічної травматизації).
Сучасні уявлення щодо ролі центральних відділів симпатичної нервової системи базується на даних про адренергічні рецептори ЦНС. З’ясовано, що різні структури мозку, які мають α -адренорецептори, контролюють активність симпатичних нервів на периферії, а отже і рівень АТ. Особливе значення надається специфічним вузлам гіпоталамуса і ділянці Nucleus Tractus Solitarii (NTS) довгастого мозку, де розташовані первинні синапси синоаортальних барорецепторних нервів, є багато норадренергічних нейронів з високою концентрацією норадреналіна.
Норадреналін, стимулюючи α 2-адренорецептори гіпоталамуса і ділянки NTS довгастого мозку, викликає нисхідне гальмування еферентної симпатичної активності і в такий спосіб сприяє розширенню судин на периферії і зниженню АТ. В разі ж дефіциту мозкового норадреналіну зростає кількість норадреналіну на периферії - в системній циркуляції. Таким чином, порушується рівновага між мозковим і периферійним норадреналіном в сторону зменшення першого і збільшення другого. Підтвердженням цієї концепції є очевидний гіпотензивний ефект клофеліна, гуанацина і гуанабенза, які стимулюють α -адренорецептори норадренергічних нейронів головного мозку, внаслідок чого зменшується потік судинозвужувальних імпульсів на периферію.
Новітні дослідження стверджують, що у ростральному відділі вентролатеральних ділянок довгастого мозку є І-імідазолінові рецептори першого і другого типів, які поряд з α 2-адренорецепторами, відіграють важливу роль у центральній регуляції АТ. На цій основі синтезовані агоністи імідазолінових рецепторів - першого (фізіотенз, моксонідин) та другого типів (рилменідин).
Ще одним підтвердженням ролі симпатичної нервової системи в розвитку ГХ є доведений факт наявності гіперадренергічної форми, яка спостерігається приблизно у 40% хворих на ГХ. У них середня концентрація катехоламінів у крові більше, як у два рази перевищує нормальний їх рівень.
Доведено також, що крім класичних медіаторів симпатичні нейрони синтезують і виділяють в кров біологічно активні пептиди, серед яких дуже важливим є нейропептид Y (NPY), який складається з 36 амінокислот. Центральний NPY локалізується в норадренергічних нейронах NTS і в допамінергічних нейронах гіпоталамуса. На периферії NPY депонується разом з норадреналіном в закінченнях симпатичних нервів у великих і щільних міхурцях. Поряд з норадреналіном нейропептид NPY сприяє підвищенню ЗПСО і АТ.
Як уже вказувалось, в мозковій тканині знайдені всі елементи системи “ангіотензиноген-ренін-ангіотензин ІІ”. Ця система функціонує синергічно з нирковою. При її домінуванні формується ангіотензин ІІ-залежна клінічна форма ГХ, яка протікає з впертою діастолічною гіпертензією. Таким хворим показані інгібітори АПФ (капотен, еналаприл), а також антагоністи рецепторів ангітензина ІІ (лозартан, ірбезартан). Власне іноді лише поєднання цих двох груп препаратів (еналаприл+ірбезартан) дає можливість домогтися нормалізації АТ.
Узагальнюючи роль депресорних механізмів у розвитку ГХ слід відмітити, що у всіх випадках пригнічення синтезу ниркою депресорних речовин (пошкодження медули нирок) має місце так звана ренопривна артеріальна гіпертензія. При ішемії нирок розвивається ренопаренхіматозна артеріальна гіпертензія.
Дуже важливою для лікаря є розуміння концепція “сольової гіпертензії”. Ниркову теорію походження ГХ у свій час визнавали Є.М.Тареєв, Г.Гольдблат, А.Гайтон. Так, Гольдблат ще в 1934 році довів, що звуження ниркової артерії і пов’язане з цим посилене виділення ішемічною ниркою реніна викликає підвищення АТ, а отже може бути причиною ГХ. Згідно концепції А.Гайтона, яка найбільш об’єктивно трактує механізм “тиск-натрійурез”, в нормі сам по собі артеріальний тиск здійснює вирішальний вплив на величину натрій- і гідруреза. При АТ біля 100 мм рт.ст. забезпечується виділення натрію і води, адекватне їх поступленню в організм. Якщо АТ підвищується до 150 мм рт.ст., то натрій- і гідрурез зростають у 3 рази. Від’ємний баланс натрію і води зберігатиметься до тих пір, поки АТ не понизиться до 100 мм рт.ст. Якщо АТ знижується до рівня < 100 мм рт.ст., виділення нирками натрію і води зменшується, а при АТ 50 мм рт.ст. – припиняється.
У хворих на ГХ цей механізм пошкоджений, тобто для видалення з сечею рівновеликої (в порівнянні із здоровими людьми) кількості натрію і води потрібний більш високий АТ.
Серед внутрішньониркових ненормальностей, які обмежують функціонування механізму “тиск-натрій-гідрурез”, тобто зменшення екскрекції натрію і води при підвищенні АТ, найважливішими є: посилений вплив симпатичної нервової системи з надмірним утворенням АІІ, спадкові дефекти медули нирок. У людей з дефектом мозкової речовини нирок розвивається напружений сольовий режим, який в молодості ще врівноважується, але з віком стає надмірним. Не випадково ця гіпергідратаційна (об’єм-, натрій-залежна) форма ГХ реєструється частіше у осіб 45-50 років. Особливо це торкається жінок в період статевої інволюції.
Розмаїття клінічних форм ГХ не обмежується лише поданими вище гіперадренергічною, ангіотензин ІІ-залежною, гіпергідратаційною (об’єм-, натрій-залежною) формами цього захворювання. Пошуки можливих причин звуження резистивних судин привели до кальцієвого механізму формування ГХ. Це так звана мембранна теорія походження ГХ (С.М.Орлов, Ю.В.Постнов). У хворих з кальцій-залежною клінічною формою ГХ спостерігається підвищений вміст Са++, зв’язаного з мембранами внутрішньоклітинних органел. З підвищенням концентрації вільного Са++ в цитоплазмі зв’язують підвищену скоротливість гладких м’язів судинної стінки, що призводить до зростання ЗПСО і підвищення АТ. Концентрація іонів Са++ в плазмі у таких хворих, навпаки, знижена. Цим і характеризується “кальцієвий парадокс”: багато іонів Са++ в гладком’язових клітинах судинної стінки і менше, ніж в нормі у плазмі. Доведено, що при дефіциті кальцію в плазмі крові в гемоциркуляції з’являється специфічний гуморальний пептид – РТН, який сприяє посиленому входженню іонів Са++ в клітини. Цей пресорний пептид (РТН) виділяють паращитовидні залози, він не ідентичний відомому паратгормону, відрізняється він і від дигіталісоподібного фактора, як і від вазопресина, АІІ, катехоламінів. У відповідь на вживання препаратів кальцію АТ у таких хворих знижується, що є підтвердженням реальності існування кальцій-залежної форми ГХ.
Серед сучасних концепцій патогенезу ГХ певне значення має й церебро-ішемічна форма цього захворювання. Встановлено, що порушення оксигенації нервових центрів спричиняють виникнення застійних вогнищ збудження у корково-підкіркових ділянках ЦНС, відповідальних за формування вазоконстрикторної домінанти. Так формується систоло-діастолічна гіпертензія у людей похилого віку, зумовлена хронічною помірною ішемією деяких ділянок мозку. За даними Д.Ф.Чеботарьова (1978) така форма зустрічається у осіб старших 80 років у 16-18% випадків.
Що торкається спадкової схильності до захворювання на ГХ, то, очевидно, вона носить полігенний характер з переважанням активності різних генів у різних людей.
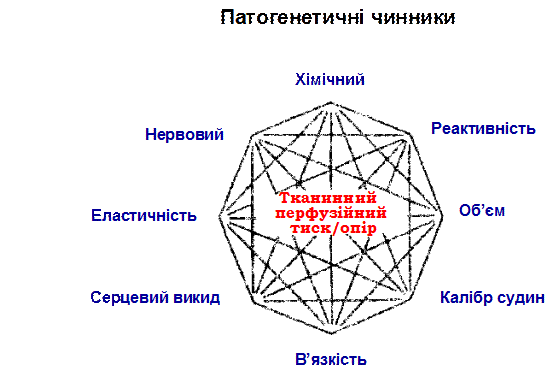
Рис. 11. Восьмикутник І.Page, який характеризує " мозаїчну"
теорію походження гіпертензивної хвороби.
Особливої уваги заслуговує “мозаїчна теорія” гіпертензивної хвороби І.Пейджа, в якій патогенез ГХ порівнюється зі складною мозаїкою, яка змінюється, як у калейдоскопі. Особливістю ГХ є те, що її початкові, пускові механізми з розвитком хвороби немов би сходять зі сцени (“Мавр зробив свою справу, мавр може відійти”), стають ледь помітними і те, що лікар бачить у хворого з 10-20-річним анамнезом - це релікти процесів, які вже “відзвучали”, залишки колись активних механізмів, “сліди метеоритів на поверхні планети”.
Визначаючи обов’язкову і значну роль симпатичної нервової системи і нирок, лікарям слід дотримуватись погляду про поліетіологічність і поліпатогенетичність гіпертонічної хвороби. Це водночас “хвороба регуляції” і “хвороба компенсації” за М.С.Кушаковським (1995).
Класифікація. Клініка. Насамперед є важливо з’ясувати деякі термінологічні особливості різних станів, що проявляються підвищенням артеріального тиску крові - м’яка, помірна, важка, ізольована систолічна, межова гіпертензія тощо (табл. 15).
| Таблиця 15. | ||
| Класифікація артеріальної гіпертензії залежно від рівня артеріального тиску, адаптована з рекомендаціями Об’єднаного Національного Комітету США (JNCV-V Report of Joint Nationalee Commitee USA, 1999) | ||
| Категорія | Систолічний артеріальний тиск, мм рт.ст. | Діастолічний артеріальний тиск, мм рт.ст. |
| Нормальний артеріальний тиск | < 130 | < 85 |
| Високий нормальний артеріальний тиск | 130-139 | 85-89 |
| Артеріальна гіпертензія: Стадія І (м’яка) Стадія ІІ (помірна) Стадія ІІІ (важка) Стадія ІV (дуже важка) | 140-159 160-179 180-209 > 210 | 90-99 100-109 110-119 > 120 |
Термін “м’яка” гіпертензія (140-159/90-99 мм.рт.ст.) використовується для характеристики ступеня підвищення артеріального тиску. В той же час абсолютний ризик серцево-судинних захворювань у хворого може бути і не “м’яким”. Термін “помірна” (160-179/100-109 мм рт.ст.) та “важка” гіпертензія (180-209/110-119 мм рт.ст.) відображають більш виражене підвищення артеріального тиску, але він так само, як і “м’яка” гіпертензія не характеризує величину загального ризику хворого.
Термін “ізольована систолічна гіпертензія” характеризує тих хворих, у яких рівень систолічного артеріального тиску понад 140 мм рт.ст., а діастолічний артеріальний тиск – менше 90 мм рт.ст.

Рис. 12. Ураження органів-мішеней при артеріальній гіпертензі
Окремо виділяють так звану “межову” артеріальну гіпертензію (140-159/90-94 мм рт.ст.). Для таких пацієнтів характерно, що нормалізація артеріального тиску відбувається спонтанно, без впливу гіпотензивних засобів, після відпочинку. У них виключена можливість симптоматичної (вторинної) гіпертензії, відсутні ознаки гіпертрофії лівого шлуночка (за даними ЕКГ, ЕхоКГ), немає змін на очному дні, нормальна функція нирок. Характерне фізіологічне добове коливання артеріального тиску при його моніторуванні, відсутні нічні підйоми артеріального тиску.
Межова артеріальна гіпертензія зустрічається не лише у молодому віці (юнацька межова артеріальна гіпертензія), але й у осіб старших вікових груп. Вона спостерігається у 10% населення. У віці до 50 років частіше є у чоловіків, а після 50 років - у жінок. Межова гіпертонія не є нозологічною одиницею, це дисрегуляторний стан організму. В третині випадків цей стан трансформується в гіпертонічну хворобу. Є декілька клінічних варіантів межової артеріальної гіпертензії, основними з яких є: 1) юнацька межова артеріальна гіпертензія (часто протікає на фоні нейроциркуляторної дистонії); 2) “клімактерична” межова артеріальна гіпертензія; 3) “алкогольна” межова артеріальна гіпертензія; 4) межова артеріальна гіпертензія у спортсменів; 5) межова артеріальна гіпертензія внаслідок тривалого впливу професійних шкідливостей (шум, вібрація, інтоксикація) – так званий “професійний” варіант.
Межова артеріальна гіпертензія в діагнозі вказується поряд з основним захворюванням. Наприклад: “Нейроциркулятона дистонія з межовим типом артеріальної гіпертензії.” До артеріальних гіпертензій відносяться есенціальна гіпертензія або гіпертензивна (гіпертонічна) хвороба (первинна артеріальна гіпертензія) і вторинні артеріальні гіпертензії (ниркові, ендокринні, внаслідок ураження мозку, аорти та магістральних її гілок, медикаментозні тощо). Отже, термін “артеріальна гіпертензія” є загальним, під ним розуміють будь-яке підвищення артеріального тиску незалежно від причини, які привели до цього.
Тривалий час захворювання перебігає без жодних клінічних ознак. Артеріальну гіпертензію знаходять випадково при обстеженні чи диспансеризації. Інші хворі відчувають підвищення кров’яного тиску підчас або після фізичних чи психоемоційних навантажень. Власне в цей період спостерігається надмірна судинна збудливість. Артеріальний тиск підвищується у цих хворих при зануренні китиць рук у холодну воду (4˚ С) або при гіпервентиляційній пробі. Прийом 2 краплин або 1/2 таблетки нітрогліцерину під язик різко знижує систолічний і діастолічний артеріальний тиск. Таких осіб називають “гіперреакторами”. У частини з них в подальшому розвивається гіпертензивна хвороба. Згодом хворих починає турбувати головний біль, який може тривати упродовж декількох днів або й місяців. Особливо виснажливим є тупий головний біль зранку після сну. Його локалізація може бути різною: в потилиці, скронях, лобній чи тім’яній ділянках. Головний біль залежить, очевидно, від подразнення рецепторів судин: він часто носить характер мігрені, або виникає у вигляді нападу, триває упродовж багатьох годин і закінчується блюванням. Іноді ж головний біль протікає по типу церебрального ангіогіпотічного кризу з ознаками набряку мозку.
Частими при гіпертензивній хворобі є запаморочення і шум у вухах. Запаморочення є наслідком порушення тонусу судин і розладу церебрального кровообігу. Хворим здається, що земля “втікає з-під ніг”, а будинки, різні вивіски й реклами “крутяться, неначе карусель”. Спазм мозкових судин проявляється шумом в голові, який спочатку є не постійним, в подальшому він набуває постійного характеру (склерозування судин). Хворі подають також скарги на швидку втому, безсоння, підвищену психічну збудливість, серцебиття, тривалий біль в ділянці серця (верхівки), неможливість спати на лівому боці. Характерний симптом “мертвого пальця”– раптова блідість одного або декількох пальців без усякої на те явної причини (іноді ця ознака з’являється після охолодження, занурення рук у холодну воду). Частими ознаками є оніміння й похолодання кінцівок, відчуття “пересування мурашок по шкірі”.
Біль в ділянці серця спочатку носить характер кардіалгії, але відтак, коли приєднується атеросклероз вінцевих артерій, розвивається ІХС з нападами стенокардії напруження, спокою.
При об’єктивному обстеженні хворих часто знаходять надмірну масу тіла, гіперемію шкіри (hyperthensia rubra). Це гіперадренергічна форма гіпертонічної хвороби. У інших хворих внаслідок спазму шкірних судин покрив стає блідим (hyperthensia alba). При блідій гіпертонії за F.Volhard капіляроскопія вказує на звуження артеріол, капілярів і дрібних вен, що є наслідком надмірного викиду ангіонтензину ІІ. Це спостерігається при ангіотензин ІІ-залежній формі гіпертензивної хвороби. Пульс на променевій артерії напружений, ритмічний. При вираженому атеросклерозі променеві та плечові артерії стають тверді, звивисті, пульсуючі.
При огляді ділянки серця знаходять посилений верхівковий поштовх, який припіднімає передню стінку грудної клітки, він зміщений вліво і вниз. Пальпаторно можна визначити акцент ІІ тона на аорті, а також пульсацію розширеної аорти в яремній ямці. При аускультації та фонокардіографії відзначають високий (акцентований) ІІ тон та грубий систолічний шум на аорті. Іноді на аорті внаслідок відносної недостатності аортального клапана спостерігається протодіастолічний шум. На верхівці внаслідок відносної недостатності мітрального клапана знаходять систолічний шум в поєднанні з послабленим І тоном.
Рентгенографія грудної клітки. В ранніх стадіях (концентричної гіпертрофії) виявляють лише заокруглену тінь верхівки лівого шлуночка. При більш вираженому, але все ж ще помірному збільшенні розмірів лівого шлуночка верхівка серця опускається дещо вниз. Якщо ж розміри лівого шлуночка продовжують збільшуватись, то верхівка зміщується вліво, зменшуючи просвіт нижнього легеневого поля. При цьому у другому косому положенні можна спостерігати звуження ретрокардіального простору (за рахунок збільшення лівого шлуночка та лівого передсердя), талія серця дещо згладжується. В більш пізніх стадіях захворювання відбувається збільшення всіх відділів серця: правий шлуночок розширюється вправо, відтісняючи догори і дозаду праве передсердя. За рахунок збільшеного правого шлуночка звужується ретростернальний простір. Поперечник серця збільшується, воно набуває трикутної форми.