Главная страница Случайная страница
КАТЕГОРИИ:
АвтомобилиАстрономияБиологияГеографияДом и садДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеталлургияМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРелигияРиторикаСоциологияСпортСтроительствоТехнологияТуризмФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Розчинники
|
|
Вступ.
2.Методи дослідження доброякісності лікарських речовин.
2.1 Хімічні кількісні методи аналізу лікарських речовин.
2.1.1 Гравіметричний Аналіз
2.1.2 Титрометричні методи аналізу
2.1.3 Методи осадження. Аргентометрія
2.1.4 Комплекосонометрія
Кислотно-основне титрування
2.1.6 Методи окислення-відновлення
2.2 Фізичні і фізико-хімічні методи дослідження лікарських засобів.
2.2.1 УФ-абсорбційна спектроскопія
2.2.2 Оптична спектроскопія
2.2.3 ІЧ-спектроскопія
2.2.4 Спектроскопія комбінаційного розсіювання світла
ЯМР
2.2.6 Мас-спектроскопія
2.2.7 Рентгеноструктурний та рентгеноспектральний аналіз
2.2.8 Термогравіметричний аналіз
2.2.9 Рефрактометрія
2.2.10 Поляриметрія
2.3 Методи визначення чистоти лікарських речовин.
2.3.1 Визначення каламутності та забарвленості
2.3.2 Визначення кислотності, лужності та рН середовища
2.3.3 Випробування на граничних вміст домішок
3. Хроматографія.
3.1 Історія
3.2 Класифікація хроматографічних методів
3.3 Найрозповсюдженіші техніки Хроматографії
3.3.1 Тонкошарова хроматографія
3.3.2 Високоефективна рідинна хроматографія (ВЕРХ).
3.3.3 Адсорбційна хроматографія
3.3.4 Афінна хроматографія
3.3.5 Ексклюзійна хроматографія
Висновки.
Вступ
Визначенням доброякісності лікарських засобів займається наука Фармацевтична хімія – наука вивчає способи одержання лікарських речовин, їх будову і дією на організм, фізичні та хімічні властивості лікарських речовин, стабільність, а також методи контролю їх якості.
Методи контролю якості або фармацевтичний аналіз, займає провідне місце при вивченні фармацевтичної хімії, має свої специфічні особливості, що значною мірою відрізняють його від інших методів аналізу.
По-перше, це пов'язано з тим, що лікарські засоби мають різну хімічну природу, до їх складу можуть входити декілька речовин, у багатьох випадках хімічна будова їх дуже складна (наприклад органічні сполуки), декі групи лікарських речовин є малостійкими при зберіганні у невідповідних умовах.
По-друге, до фармацевтичного аналізу висуваються високі вимоги, критеріями яких є точність, специфічність, чутливість. Це викликано великою відповідальністю за результати аналізу, за якими стоїть життя людини – хворого, який вживатиме ліки.
Названі особливості фармацевтичного аналізу потребують як ґрунтовного знання загальних методів аналізу, так і переходу від них до багатьох конкретних методик його проведення. Разом із тим серед них є чимало специфічних.
1. Методи дослідження доброякісності лікарських речовин.
Методи дослідження якості лікарських засобів можна поділити на фізичні, фізико-хімічні та хімічні. До фізичних методів аналізу належать: визначення розчинності, прозорості і ступеня каламутності, ступеня забарвлення рідин, температури плавлення, температурних меж перегонки, показника заломлення, питомого обертання тощо. Фізичні властивості речовин в багатьох випадках служать критеріями їх чистоти.
Фізико-хімічні методи аналізу мають ряд переваг перед фізичними методами. Вони характеризуються високою чутливістю, точністю і відтворюва-ністю результатів. Фізико-хімічні методи аналізу поділяють на такі основні групи:
1. Оптичні (фотоколориметрія, УФ-, ІЧ-спектроскопія, атомно-емісійна спектрометрія, атомно-абсорбційна спектрометрія).
2. Електрохімічні (потенціометрія, амперометрія, кондуктометрія та ін.).
3. Хроматографічні (адсорбційна, тонкошарова, газова, рідинна хроматографія).
Використання цих методів дозволяє отримати додаткові характеристики досліджуваних речовин.
Фармакопея вміщує статті “Розчинність” та “Визначення густини за допомогою ареометра або пікнометра”. Уміння користуватися встановленими константами і застосовувати їх при фармакопейному аналізі дає можливість визначувати одну з характеристик якості лікарського засобу.
Одним з оптичних методів аналізу є рефрактометрія. Використання методу рефрактометрії для дослідження чистоти, ідентифікації і кількісного визначення, а також для аналізу двох- і трикомпонентних лікарських форм без попереднього розділення, підтверджує актуальність застосування цього методу.
Значна кількість органічних лікарських засобів є оптично активними речовинами. Поляриметричний метод аналізу базується на властивості оптично активних речовин відхиляти площину поляризації при проходженні крізь них прямолінійного поляризованного світла. Поляризацію використовують для ідентифікації, дослідження чистоти, кількісного визначення лікарських засобів.
За допомогою потенціометричного методу аналізу можна визначати pH розчинів, з метою встановлення доброякісності, а також для визначення кількісного вмісту лікарських засобів у їх розчинах.
2.1 Хімічні кількісні методи аналізу лікарських речовин
До методів кількісного аналізу належить:
-Фізичні методи аналізу лікарської речовини
-хімічні методи аналізу лікарської речовини
2.1.1 Гравіметричний Аналіз
Гравіметричний аналіз базується на визначенні маси речовини відомого складу, утвореної або вилученої з досліджуваного лікарського засобу.
Для кількісного визначення лікарських речовин найчастіше застосовують метод преципітації (рідше методи відгонки або екстракції), який ґрунтується на осадженні речовини, що визначається, у вигляді малорозчинної сполуки відомого складу з подальшим фільтруванням отриманого продукту.
Гравіметричне визначення складається з трьох експериментальних стадій:
- Зважування наважки
- Отримання вагової форми речовини в результаті преципітації і фільтрування, екстракції чи відгонки;
- Доведення до постійної маси і зважування продукту відомого складу, отриманого з наважки.
На базі цих даних можна розрахувати вміст речовини, що визначається, у відсотках:
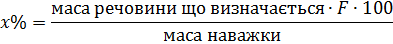
Головною перевагою гравіметричного методу аналізу є те що це метод прямого визначення, який забезпечує високу точність. Хоча в кожному окремому випадку ступінь точності залежить від багатьох факторів – розчинності осаду, спільного осадження та інше. Найбільш точним гравіметричний метод є у випадку коли вміст речовини понад 1 %. Помилка у цьому випадку складає 0.2 – 0.4 %.
До недоліків методу треба віднести велику клопітливість і тривалість проведення аналізу, що пов’язано з необхідністю використання таких операцій, як фільтрування, промивання осаду, висушування та доведення до постійної маси, а також значно нижчу селективність у випадку аналізу багатокомпонентних сумішей, що погіршує точність визначення.
Гравіметрія застосовується в аналізі таких лікарських речовин:
-натрію сульфату
-солі хініну
-солібензилпеніциліну
-прогестерон
-метиладростендіол
В сучасній аналітичній документації гравіметричний метод використовується все рідше.
Наприклад: Визначення Хініну Гідрохлориду
Осаджують основу хініеу за допомогою розчину натрію гідроксиду:
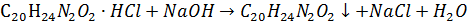
Основу хіхіну екстрагують за допомогою хлороформу; хлороформний шар відділяють, хлороформ відганяють. Залишок Висушують та зважують.
Розраховують кількісний всміст хініну гідрохлориду у відсотках за формулою:
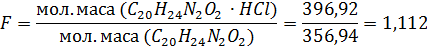
2.1.2 Титрометричні методи аналізу
Титрометричний аналіз базується на визначенні кількісного вмісту речовини за кількістю використаного стандартного розчину. В Фармацевтичному аналізі застосовується найбільш широко, оскільки це методи прямого визначення (на відміну від референсних фізико-хімічних методів, які вимагають стандартних зразків речовин, що визначаються), вони не потребують великих затрат часу, зручні та забезпечують достатньо високий ступінь точності. Для титрування використовують розчин, виготовлений шляхом розведення точної наважки безпосередньо в колбі для проведення аналізу
Титр розраховують за формулою:
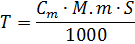
За способом проведення розрізняють методи прямого, зворотного і непрямого титрування
Пряме титрування ґрунтується на безпосередньому вимірюванні об’єму титрованого розчину, витраченого на взаємодію з речовиною, що визначається.
Розрахунок проводять за формулою:
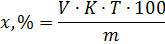
Зворотне титрування застосовують у випадках, коли реакція між досліджуваною речовиною та титрованим розчином проходить повільно, при визначенні летких речовин, у випадку застосування титрованих розчинів, концентрація яких може змінюватися при зберіганні. При зворотному титруванні фігурують 2 об’єми.
Розрахунок вмісту речовини проводять за формулою:
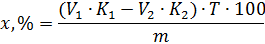
Непрямі методи титрування застосовують для речовин які не можуть кількісно прореагувати з титрованим розчином. При непрямих методах титрування відтитровують продукт, який виділяється в еквівалентній кількості при взаємодії досліджуваної речовини з будь-яким реактивом.
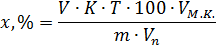
До титрометричних методів належать:
2.1.3 Методи осадження. Аргентометрія
Методи осадження ґрунтуються на утворенні при титруванні малорозчиних речовин, які випадають в осад. Для кількісних розрахунків за цими методами необхідно визначити об’єм титранту, який витрачається на повне осадження досліджуваної речовини.
Осад повинен відповідати таким вимогам:
1. Осад має бути практично нерозчинним.
2. Утворення осаду має відбуватися швидко.
3. Реакції осадження мають перебігати кількісно у відповідності зі стехіометрією хімічного рівняння.
4. Має бути можливість вибору індикатора до відповідної реакції осадження.
5. На результати титрування не повинні впливати явища адсорбції.
Найбільшою мірою цим умовам відповідають реакції утворення осадів ряду аніонів з солями ряду аніонів з солями аргентуму, на яких ґрунтується метод аргентометрії. За цим методом визначають хлорид-, бромід-, тіоціанід-іони.
Наприклад: Визначення хлоридів (індикатор калію хромат, забарвлення цегельно червоний осад аргентуму хромату).
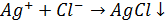
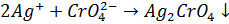
Умови титрування: середовище близьке до нейтрального (pH 6, 3-10, 0).У кислому середовищікалію хромат перетворюється у дихромат:
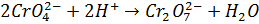
При pH> 10, 0 можливе протікання реакції:
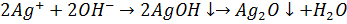
1.1.4 Комплекосонометрія
В аналітичній практиці широке застосування знаходить компексонометрія- титрометричний метод аналізу, який ґрунтується на утворенні стійких комплексних сполук катіонів полівалентних металів з різноманітними лігандами. До комплекксиметрії належить меркуриметрія – титрування солями 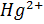 , меркурометрія – титрування солями
, меркурометрія – титрування солями 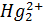 .
.
Точку еквівалетності у комплексонометричному титруванні можна встановити за допомогою фізичних методів (потенціометрично, амперометричний) однак на практиці відають перевагу візуальному індикаторному способу, як найбільш простому, зручному та швидкому. Індикатори, які використовують у комплексонометрії, називають металоіндикаторами. Метало індикатори – це органічні барвники які утворюють з іонами металів інтенсивно забарвлені комплекси.
При цьому більшість індикаторів приєднують або віддають протони, змінючи при цьому забарвлення.
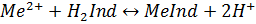
1.1.5 Кислотно-основне титрування
Метод кислотно-основного титрування ґрунтується на реакціях кислотно-основної взаємодії, які в загальному вигляді можна представити таким чином:
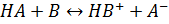
Сила кислоти або основи значною мірою залежить від кислотно-основних властивостей розчинника. Розчинення багатьох речовин можна представити як реакції утворення супряжених кислот і основи:
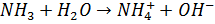
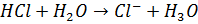
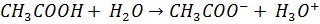
Сила кислоти у водному розчині визначається тим, наскільки повно вона віддає протони молекулам води, а сила онови – наскільки вона акцептує протони молекул води.
Кислотно основне титрування включає в себе Алкаліметрію в водном і безводном середовищі
В водном визначення HCls=1
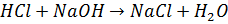
Ацидиметрія ацидиметричні методи титрування у фармацевтичному аналізі використовують для визначення органічних основ або солей утворених сильними основами та слабкоми кислотами. Неподілена електрона пара нітрогену зазвичай підбирають індикатор з переходом забарвлення в кислому середовищі: метиловий оранжевий, метиловий червоний.
Визначення аміаку розчину конц.
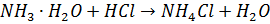
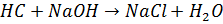
S=1Зворотна ацидиметрія Індикатор – який має перехід в слабкокислому середовищі, що відповідає рН гідролізу продукту реакції- Амонію хлориду.
1.1.6 Методи окислення-відновлення
Методи окисно-відновного титрування ґрунтуються на використанні процесів, повязаних з переносом електронів, тобто окисно-відновних реакцій
Це дуже розповсюджені методи титриметричного аналізу, що дозволяють прямо або зворотно визначати неорганічні лікарські речовини, здатні за певних умов стехіометрично приймати або відавати елекрони, тобто бути окиснені до речовин з менш ступенем відновлення, ніж віхідні речовини.
У методах окисно-відновного титрування використовують пряме, зворотне, та титрування за замісником.
Пряме титрування використовують у тих випадках коли швидкість реакції досить швидко.
Зворотне титрування використовують, якщо реакція перебігає повільно та для її завершення потрібний час, а також для визначення летких сполук та тих, що прямо не реагують з титрантом.
Титрування за замісником використовують у тих випадках, коли досліджувана речовина не реагуює з титрантом або реакція перебігає не стехіометрично.
Кінцеву точку титрування в окисно –відновних методах визначають безідикаторним методом або за допомогою специфічних та редок- індикаторів.
-Перманганатометрія
Метод грунтується на застосуванні реакцій окиснення лікарської речовини, що визначається, перманганат іонами. Продукти відновлення перманганат-іонів можуть бути різними залежно від рН середовища:
- Усильно кислому середовищі:
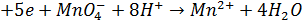
- У слабкокислому або нейтральному середовищі:
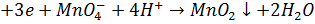
- У слабколужном середовищі:
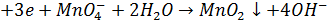
Для створення кислого середовища застосовують сульфатну кислоту. Даний метод є безіндикаторним титрування проводять до знебарвлення розчину. Визначають Перманганатометрично Розчин водню пероксиду 3%
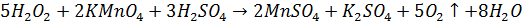
Йодометрія
Йодометрія – метод кількісного визначення вільного йоду або тих речовин, які кількісно виділяють його під час реакцій, і тих сполук, які зв’язують йод або окислюються йодом у стехіометричних кількостях.
Йодометричний метод кількісного визначення має широке ппрактичне застосування; за своєю природою і точністю він визнається одним із кращих редокс-методів кількісного визначення.
В основі йодометричного визначення лежать реакції
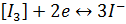
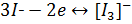
Йод здатний вступати в реакції приєднання, які використовується в аналітичній хімії для визначення подвійних зв’язків у не насичених органічних сполуках, а також в реакції заміщення атомів гідрогену в ароматичних та гетероциклічних сполуках.
Кінцеву точку титрування в йодометрії визначають:
А) без індикатора – калій йодит забарвлений у бурий колір, тому при титруванні надлишкова капля забарвлює розчин
Б) з індикатором - крохмаль
Також сюди належить:
-Йодохлорометрія
-Йодатометрія
-Броматометрія
- Нітритометрія
- Цериметрія
- Дихроматометрія
2.2 Фізичні і фізико-хімічні методи дослідження лікарськіх засобів
Фізичні методи аналізу — сукупність методів дослідження структури, складу та властивостей речовин, а також фізико-хімічних процесів, що в них відбуваються, з метою їх ідентифікації та створення нових речовин із заданими властивостями.
Усі Ф.м.а. можна класифікувати як за характером взаємодії речовини з різноманітними видами зовнішніх впливів, так і за тими властивостями речовини, які можна цими методами визначити. Речовину опромінюють усією сукупністю електромагнітних хвиль від гамма-квантів до радіодіапазону, а також ультразвуком у широкому діапазоні аж до надвисоких частот (гігагерци), поміщають у сильні постійні електричні та магнітні поля, бомбардують електронами, протонами, нейтронами, а також атомними та молекулярними пучками, нагрівають до високих і охолоджують до наднизьких температур аж до 10–3 К і нижче, використовують глибокий вакуум і надвисокий тиск, а також різні комбінації цих впливів.
При класифікації Ф.м.а. за типом досліджуваних систем і їх властивостями враховують методи визначення структури речовини, зокрема, геометричної будови молекул, електронних, коливальних і обертальних енергетичних спектрів, електричних дипольних моментів, ат. і мол. м. та ізотопного складу речовини тощо. Для дослідження речовини та одержання інформації щодо процесів, які у ній виникають, придатний будь-який зовнішній вплив, як хімічний, так і фізичний, аби досліджувана система на нього відгукувалася, і цей відгук, захоплюючи та передаючи необхідну інформацію про речовину, піддавався б однозначному розумінню. На відміну від хімічних, Ф.м.а. мають набагато більшу точність і чутливість.
У свою чергу, дослідження взаємодії електромагнітних хвиль різноманітного діапазону з речовиною (спектральні методи аналізу) розподіляють на піддіапазони залежно від того, з якою з чотирьох квантових підсистем молекули взаємодіє випромінювання, яке потрапляє на речовину: з ядерною системою — гамма-кванти (гамма-резонансна або месбаурівська спектроскопія) та електромагнітні хвилі радіочастотного НВЧ-діапазону (спектроскопія ЯМР); з електронною системою атомів і молекул — рентгенівське, УФ-, видиме та ближньої ІЧ-області випромінювання (рентгенівська та оптична спектроскопія); з коливальною системою атомів у молекулі — ближня і середня ІЧ-область (коливальна спектроскопія); з молекулярною (як ціле) обертальною системою — дальня ІЧ-область (обертальна спектроскопія).
Сучасні варіанти спектрального аналізу використовують ефект поглинання електромагнітних хвиль речовиною і називаються відповідно абсорбційними або спектрами поглинання, більш чутливими, одержаними в більш контрольованих умовах порівняно зі спектрами випромінювання. Взаємодія пучків електронів, нейтронів, атомів, а також рентгенівських квантів з речовиною, яка спричиняє їх розсіювання та утворення відхилених (дифрагованих) пучків, спрямованих під певними кутами до первинного пучка, є основою так званих дифракційних методів аналізу.Для аналізу та ідентифікації речовин, зокрема, лікарських, застосовують в основному такі Ф.м.а.:
2.2.1 УФ-абсорбційна спектроскопія — один із розділів оптичної спектроскопії, що базується на отриманні та дослідженні спектрів поглинання в УФ-області спектра (діапазон довжин хвиль ∆ λ: 190/400 нм; λ < 190 нм — вакуумна УФ-область малопридатна для роботи через сильне поглинання хвиль повітрям). Поглинання УФ-випромінювання зумовлене електронними переходами в атомах з основного енергетичного стану в більш високий (збуджений); у молекулах — зі зв’язувальної орбіталі (основний стан) на розпушувальну орбіталь (збуджений стан).
2.2.2Оптична спектроскопія (видимий діапазон, ∆ λ: 400/700 нм) базується, як і УФ-спектроскопія, на квантових переходах між електронними енергетичними рівнями атомів і молекул.
2.2.3 ІЧ-спектроскопія
ІЧ-спектроскопія — розділ оптичної спектроскопії, який базується на отриманні та дослідженні спектрів поглинання в ІЧ-області спектра (∆ λ: 1/2, 5 мкм — ближня, ∆ λ: 2, 5/50 мкм — середня і ∆ λ: 50/300 мкм — дальня ІЧ-область спектра). Поглинання ІЧ-випромінювання зумовлене енергетичними переходами у квантовій коливальній системі атомів у молекулі (в основному, середня ІЧ‑ область), а також квантовими переходами в обертальній енергетичній системі молекули (дальня ІЧ-область). ІЧ‑ спектр — сукупність смуг поглинання, їх положення та інтенсивність — характерний для даної речовини і дозволяє визначити молекулярну структуру речовин та їх хімічний склад.
2.2.4 Спектроскопія комбінаційного розсіювання світла
Спектроскопія комбінаційного розсіювання світла (КРС-спектроскопія або Раман-ефект) базується на розсіюванні світла молекулами, яке супроводжується зміною частоти розсіяного світла за рахунок того, що енергія первинного світлового кванта може перевести молекулу на інші коливальні та обертальні рівні енергії порівняно з початковим. При цьому частоти нових ліній у спектрі розсіювання є комбінаціями частоти світла, що падає на речовину, і частот коливальних та обертальних переходів у молекулі (так звані стоксові та антистоксові лінії). Вимірюючи частоти цих ліній та знаючи частоту первинного випромінювання, можна визначити частоти власних (нормальних) коливань молекули, характерних для кожної речовини. Спектри КРС та ІЧ не дублюють, а доповнюють один одного, оскільки визначаються різними правилами добору і при сумісному використанні цих методів може бути одержана максимальна інформація про коливальні та обертальні спектри досліджуваної речовини.
ЯМР
ЯМР — комбінований метод дослідження структури речовини, що базується на взаємодії спінової ядерної системи з електромагнітними хвилями радіочастотного НВЧ-діапазону (λ -метри, дециметри) у сильному постійному магнітному полі. Метод полягає у вибірковому (резонансному) поглинанні енергії хвиль речовиною, зумовленому переорієнтацією магнітних моментів ядер. Найбільше поширення, зокрема, в органічній хімії, набув метод ЯМР на ядрах атома водню — протонах (ПМР — ПМР). Спектр ЯМР — сукупність сигналів переходів між енергетичними рівнями у спіновій ядерній системі — специфічний для кожної речовини і може служити для її ідентифікації як один із кращих сучасних Ф.м.а.
2.2.6 Мас-спектроскопія
Мас-спектроскопія — комбінований метод дослідження структури та властивостей речовини, що ґрунтується на іонізації та руйнуванні молекул, напр., потоком електронів, розподілі в просторі утворених фрагментів за їх масами у силових полях (електричному та магнітному) і визначенні мас цих уламків, сукупність яких та їх відносний вміст і є мас-спектром досліджуваної речовини. Структурна мас-спектроскопія — один із найбільш інформативних сучасних методів дослідження та аналізу речовини — широко використовується для визначення ат.м. та мол. м., структури молекули, ізотопного складу речовини, а також чистоти ЛП.
2.2.7 Рентгеноструктурний та рентгеноспектральний аналіз
Рентгеноструктурний та рентгеноспектральний аналіз — сукупність методів дослідження структури і складу речовини, що базується на дифракції рентгенівських променів на її атомній структурі та їх випромінюванні або поглинанні внутрішньою електронною системою атомів. На відміну від оптичних, рентгенівські спектри атомів різноманітних хімічних сполук (як простих, так і дуже складних) не змінюються тому, що внутрішні електронні енергетичні рівні атомів не деформуються і зберігають свою індивідуальність, що дозволяє застосовувати рентгенівські методи для аналізу елементного складу речовини.
2.2.8 Термогравіметричний аналіз
Термогравіметричний аналіз — фізико-хімічний метод аналізу теплових ефектів, фазових переходів і хімічних реакцій, які відбуваються в речовині при зміні її температури. Всі фазові перетворення відображаються у вигляді особливостей на кривих охолодження або нагрівання зразка — залежності температури від часу — термограмі. Кількість тепла для будь-якої хімічної реакції (екзотермічної або ендотермічної) пропорційна масі речовини, що прореагувала. Тому для аналітичних цілей великий інтерес становлять термогравіметричні методи аналізу, за допомогою яких процеси, що відбуваються в речовині, визначають за зміною маси при зміні температури на термогравіметричних залежностях m (T), які доповнюють термограми. Сучасні дослідження теплових ефектів базуються на методі диференціальної (дериватної) термогравіметрії.
2.2.9 Рефрактометрія
Рефрактометрія — оптичний метод дослідження речовин, що ґрунтується на явищі заломлення світла границею розподілу двох середовищ і вимірюванні показника заломлення речовини, який є його специфічною характеристикою. Метод використовується для визначення складу і контролю якості речовини, для визначення невідомої концентрації речовини у розчині, встановлення аутентичності та чистоти лікарських речовин.
2.2.10 Поляриметрія
Поляриметрія — оптичний метод дослідження речовини, в основу якого покладено вимірювання кута повороту площини поляризації плоскополяризованого світла, яке пройшло крізь оптично активну речовину. Метод широко застосовується для визначення концентрації оптично активних речовин у розчині, а також для оцінки їх чистоти. Питоме обертання площини поляризації є характерною сталою величиною для цієї речовини, яку можна використовувати для її ідентифікації, в т.ч. визначення ЛП.
Важливою особливістю Ф.м.а. є зміщення акцентів у бік застосування методів, де використовується комбінований вплив на речовину (ЯМР, мас-спектроскопія тощо), а також комплексне дослідження речовин, що дуже важливо для вперше синтезованих сполук.
2.3 Методи визначення чистоти лікарських речовин
Практично всі лікарські речовини мають у своєму складі стороні домішки. Їх наявність може бути зумовлена такими причинами:
- недостатньою чистотою вихідних продуктів, які застосовуються при синтезі або вилученні з сировини;
- утворення побічних продуктів у процесі синтезу;
- екстракцією сторонніх речовин із рослинної сировини;
- екстракцією катіонів металів із матеріалу реакторів або намолом робочого органу млинів;
- розкладання лікарської речовини при неправильному зберіганні або забрудненням продуктами розкладу таропакувальних матеріалів.
Залежно від характеру та властивостей домішки можуть бути поділенні на дві групи:
- домішки, які характеризують ступінь очистки препарату;
- домішки, які впливають на фармакологічну дію.
У кожній окремій фармакопейній статті наводиться перелік показників, за якими перевіряється чистота лікарської речовини(зовнішній вигляд, розчиність, температура топлення або кипіння, забарвленість розчину).
Невідповідність лікарської речовини навіть одному з показників, закладених у нормативно-технічній документації, свідчать про її недоброякісність.
Наприклад:
2.3.1 Визначення каламутності та забарвленості
Лікарські речовини можуть бути забрудненими домішками, поява яких зумовлена зміною їх фізичних та хімічних властивостей під впилов вологості, світла, кисню, вуглекислого газу та інших факторів навколишнього середовища.
Наявність домішок частіше за все викликає зміну забарвлення розчинів лікарських речовин або розчинності у воді та інших розчинниках. Контроль за прозорістю розчинів лікарських речовин регламентується вказівками загальної фармакопейної статті та монографій. Ступінь каламутності визначають порівнянням рідин або розчину, що досліджується, з розчиником або еталоном. Для приготування еталона використовують вихідну суспензію, що досліджується утворюється в результаті реакції водних розчинів гідразину сульфату та гексаметилентетраміну. Еталонів, з якими порівнюють ступінь каламутності, чотири. Випробувальну рідину вважають прозорою, якщо вона витримує порівняння з водою або розчинником, використаним при приготуванні випробувальної рідини при перегляді за описаних вище умов, або її каламутності не перевищує каламутність еталона I.
2.3.2 Визначення кислотності, лужності та рН середовища
Невідповідність лікарської речовини за цими показниками може бути наслідком присутності домішок більш кислотного або основного характеру, ніж сама речовина.
Подібні домішки можуть утворюватися при добуванні або зберіганні лікарських засобів(домішки мінеральних кислот, продуктів гідролізу, вплив лужності скла, поглинання речовиною вуглекислого газу з повітря).
Контроль кислотності, лужності, рН середовища регламентується відповідним розділом монографій та може здійснюватися різними способами:
- за зміною забарвлення кислотно- основних індикаторів (рН індикаторів)
- визначенням величини рН за методом, наведеним у загальній фармакопейній статті «Визначення рН». Таке визначення є обовязковим для всіх ін’єкційних розчинів;
- кислотно-основним титруванням.
2.3.3 Випробування на граничних вміст домішок
Державна фармакопея не вимагає абсолютної чистоти лікарського засобу і не допускає в ньому певні домішки в сурово визначених межах. У фармакопейному аналізі існують два методи визначення вмісту домішок:
Наприклад:
Дослідження на солі амонію – залежно від властивостей лікарської речовини та допустимої домішки солей амонію її визначення проводиться за одним із чотирьох методів.
Метод А грунтується на здатності солей амонію утворювати солі амонію з розчином калію тетрайодомеркурату лужного(Реактив Несслера) жовто-бурий осад або жовте забарвлення залежно від концентрації.
NH4+ + 2[HgI4]2‾ + 2OH‾ → [NH2Hg2I2]+I‾ + 5I‾ + 2H2O.
Виявлення домішки кальцію
При приготуванні всіх розчинів, застосовуваних у даному випробуванні, має використовуватися вода дистильована Р.
До 0.2 мл еталонного розчину кальцію спиртового (100 ppm Ca) Р додають 1 мл розчину амонію оксалату Р. Через 1 хв. додають суміш 1 мл кислоти оцтової розведеної Р і 15 мл розчину, що містить зазначену в окремій статті кількість випробовуваної речовини, і струшують.
Паралельно за цих самих умов готують еталон, використовуючи суміш 1 мл кислоти оцтової розведеної Р, 10 мл еталонного розчину кальцію водного (10 ppm Ca) Р і 5 мл води дистильованої Р.
Через 15 хв. опалесценція випробовуваного розчину не має перевищувати опалесценцію еталона.
Ca2+ + C2O42– → CaC2O4↓.
Виявлення домішки хлоридів
До 15 мл розчину, зазначеного в окремій статті, додають 1 мл кислоти азотної розведеної Р і виливають суміш за один раз у пробірку, що містить 1 мл розчину срібла нітрату Р2.
Паралельно за цих самих умов готують еталон, використовуючи замість 15 мл випробовуваного розчину 10 мл еталонного розчину хлориду (5 ppm Cl) Р і 5 мл води Р. Пробірки поміщають у захищене від світла місце.
Через 5 хв. пробірки переглядають на чорному фоні горизонтально (перпендикулярно до осі пробірок). Опалесценція випробовуваного розчину не має перевищувати опалесценцію еталона.
Cl‾ + Ag+ → AgCl↓.
Виявлення домішки важких металів (метод А)
До 12 мл водного розчину, зазначеного в окремій статті, додають 2 мл буферного розчину рН 3.5 Р і перемішують. Одержану суміш додають до 1.2 мл реактиву тіоацетаміду Р і негайно перемішують.
Паралельно за тих самих умов готують еталон, використовуючи замість 12 мл випробовуваного розчину суміш 10 мл еталонного розчину свинцю (1 ppm або 2 ppm Pb) Р, як зазначено в окремій статті, і 2 мл випробовуваного розчину.
Готують „сліпу” пробу, використовуючи суміш 10 мл води Р і 2 мл випробовуваного розчину. Порівняно із „сліпою” пробою еталон повинен мати світло-коричневе забарвлення.
Через 2 хв. коричневе забарвлення випробовуваного розчину має бути не інтенсивнішим за забарвлення еталона.
Інший можливий хімізм:
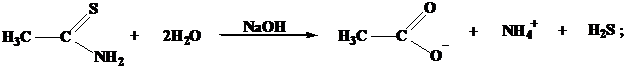
H2S → HS– + H+ → S2– + 2H+;
Pb2+ + S2– → PbS↓.
Виявлення домішки заліза
Кількість випробовуваної речовини, зазначену в окремій статті, розчиняють у воді Р, доводять об’єм розчину водою Р до 10 мл і перемішують або використовують 10 мл розчину, зазначеного в окремій статті. Додають 2 мл розчину 200 г/л кислоти лимонної Р і 0.1 мл кислоти тіогліколевої Р. Перемішують, підлужують розчином аміаку Р, доводять об’єм розчину водою Р до 20 мл.
Паралельно за цих самих умов готують еталон, використовуючи 10 мл еталонного розчину заліза (III) (1 ppm Fe) Р.
Через 5 хв. рожеве забарвлення випробовуваного розчину має бути не інтенсивнішим за забарвлення еталона.
Виявлення домішки сульфатів
При приготуванні усіх розчинів, застосовуваних у даному випробуванні, має використовуватися вода дистильована Р.
До 1.5 мл еталонного розчину сульфату (10 ppm SO4) Р1 додають 1 мл розчину 250 г/л барію хлориду Р. Струшують і залишають на 1 хв., потім додають 15 мл випробовуваного розчину, приготованого, як зазначено в окремій статті, і 0.5 мл кислоти оцтової Р.
Паралельно за цих самих умов готують еталон, використовуючи замість випробовуваного розчину 15 мл еталонного розчину сульфату (10 ppm SO4) Р.
Через 5 хв. опалесценція випробовуваного розчину не має перевищувати опалесценцію еталона.
Ba2+ + SO42‾ → BaSO4↓.
Виявлення домішки цинку
До 10.0 мл розчину випробовуваної речовини, приготованого, як зазначено в окремій статті, додають 2.0 мл розчину кислоти хлористоводневої Р1 і 0.2 мл розчину калію фероціаніду Р.
Паралельно готують еталон з використанням замість випробовуваного розчину 10 мл еталонного розчину цинк-йона (5 ppm Zn), який готують шляхом розведення водою Р у 1000 разів еталонного розчину цинку (5 мг/мл Zn) Р.
Через 10 хв. опалесценція випробовуваного розчину не має перевищувати опалесценцію еталона.
3Zn2+ + 2K+ + 2[Fe(CN)6]4‾ → K2Zn3[Fe(CN)6]2↓.
3. Хроматографія
Хроматографія — метод розділення, аналізу і дослідження сумішей речовин, що ґрунтується на різному розподілі речовин в динамічних умовах між рухомою і нерухомою фазами (на різній сорбції складових частин яким-небудь адсорбентом). Розрізняють: за середовищем, в якому відбувається розділення (газова і рідинна); за механізмом розділення (молекулярна, йонообмінна, осадова і розподільча); за технікою проведення розділення (колонкова, капілярна, тонкошарова і хроматографія на папері, HPLC (ВЕРХ, ВисокоЕфективна Рідинна Хроматографія)).Широко використовують при аналізі корисних копалин, гірських порід і мінералів у технологічних процесах для очищення і опріснення води, для отримання речовин високої чистоти. У нафтовій аналітичній практиці широко застосовуються різні види на алюмосилікатних сорбентах і на спеціальному хроматографічному папері. Інша назва — хроматографічний аналіз.
3.1 Історія
В 1850 році в роботах німецького хіміка Рунге було описано процес розділення фарбників методом фронтальної проявки на папері. Знайдені й інші роботи аналогічного характеру. Проте досліди Рунге й інших вчених не склали наукової дисципліни.
Відкрив хроматографію російський вчений Михайло Цвєт, який навчався і закінчив докторат у Женевському університеті, але наткнувся на безпрецедентне ставлення і нерозуміння в Росії.
Про хроматографічний метод він повідомив у Варшаві 21 березня 1903 р. на засіданні Варшавського товариства природодослідників; опублікував статтю в 1906 р. в журналі «Доповіді Німецького ботанічного товариства». Проте спочатку цей винахід не отримав належної оцінки і був забутий.
Відновленням інтересу до хроматографії слід завдячувати німецькому вченому Ріхардові Вільштеттеру, який запропонував використати метод Цвєта своєму учневі Ріхардові Куну. У 1931 році Р. Кун, який працював у лабораторії в Гайдельберзі провів успішне розділення каротинів на фракції. Разом з ним працювали Альфред Вінтерштайн і Едгар Ледерер. У 1933 р. Вінтерштайн продемонстрував хроматографічний метод у Кембриджському університеті, де працював молодий учений Арчер Джон Портер Мартін, а Ледерер, який у зв'язку з приходом нацистів емігрував до Парижа, і перебуваючи у 1935–1937 рр. в СРСР, познайомив із ним радянську наукову громадськість. З тих пір хроматографію почали широко використовувати в ботанічних і біохімічних лабораторіях у всьому світі.
Важливим етапом стало відкриття Миколою Ізмайловим і Марією Шрайбер методу хроматографії в тонкому шарі в 1938 році в Харківському хіміко-фармацевтичному інституті. Подальшим важливим в розвитку хроматографії стало відкриття Мартіном та Сінгом в 1940 році варіанту рідинної розподільної хроматографії на прикладі розділення похідних амінокислот на колонці, заповненій силікагелем, насиченим водою, з використанням хлороформа в якості розчинника. За це відкриття Мартін і Сінг в 1952 році отримали Нобелівську премію.
3.2 Класифікація хроматографічних методів
- Газова хроматографія (використовується наприклад для визначення якості етилового спирту)
-Рідинна хроматографія (використовується для аналізу та виділення органічних сполук)
-Хроматографія над критичними рідинами/газами (рідкісний вид, проміжний між першими двома. Найчастіше використовують як елюєнт вуглекислий газ під високим тиском та підвищених температурах)
-Газо-рідинна хроматографія
-Газо-твердофазна хроматографія
Варіанти хроматографії за способом проведення
Препаративна — використовується для розділення речовин залежно від їх швидкості руху. В більшості випадків застосовують рідинну хроматографію.
Аналітична — має на меті лиш оцінити склад суміші. Маси зразків — мікрограми.
3.3 Найрозповсюдженіші техніки хроматографії
3.3.1 Тонкошарова хроматографія
Основою тонкошарової хроматографії є адсорбційний метод, хоча також зустрічається метод розподільної хроматографії.
Адсорбційний метод заснований на розходженні ступеня сорбції-десорбції поділюваних компонентів на нерухомій фазі. Адсорбція здійснюється за рахунок ван-дер-вальсовскіх сил, що є основою фізичної адсорбції, полімолекулярної (освіта декількох шарів адсорбата на поверхні адсорбенту) і хемосорбцией (хімічної взаємодії адсорбенту і адсорбату).
Для ефективних процесів сорбції-десорбції необхідна велика площа, що висуває певні вимоги до адсорбентів. При великій поверхні поділу фаз відбувається швидке встановлення рівноваги між фазами компонентів суміші та ефективне розподіл.
Ще одним видом використовуваному в методі тонкошарової хроматографії є розподільна рідинна хроматографія.
У розподільній хроматографії обидві фази - рухлива і нерухома - рідини, що не змішуються один з одним. Поділ речовин грунтується на відмінності в їх коефіцієнтах розподілу між цими фазами.
Вперше метод тонкошарової хроматографії заявив про себе як " Паперова тонкошарова хроматографія", яка грунтувалася на розподільному методі розділення компонентів.
Розподільна хроматографія на папері.
У зв'язку з тим, що використовувана в цьому методі хроматографічна папір (спеціальні сорти фільтрувального паперу) містять в порах воду (20-22%), в якості іншої фази використовуються органічні розчинники.
Використання хроматографії на папері має ряд істотних недоліків: залежність процесу поділу від складу і властивостей паперу, зміна вмісту води в порах паперу при зміні умов зберігання, дуже низька швидкість хроматографирования (до декількох діб), низька відтворюваність результатів. Ці недоліки серйозно впливають на поширення хроматографії на папері як хроматографічного методу.
Тому можна вважати закономірним поява хроматографії в тонкому шарі сорбенту - тонкошарової хроматографії.
Основи тонкошарової хроматографії.
У методі ТШХ хроматографування речовин відбувається в тонкому шарі сорбенту, нанесеного на тверду плоску підкладку. Поділ в цьому методі в основному відбувається на основі сорбції-десорбції.
Використання різних сорбентів, дозволило значно розширити та покращити цей метод.
На початку появи методу пластини доводилося виготовляти самостійно. Але на сьогоднішній день в основному використовуються пластини заводського виготовлення, які мають досить широкий асортимент як за розмірами і носіям, так і по подложкам.
Сучасна хроматографічна пластинка являє собою основу зі скла, алюмінію або полімеру (наприклад політерефталат). У зв'язку з тим, що скляна основа стає менш популярною (часто б'ється, не можна розділити пластинку на кілька частин не пошкодивши шар сорбенту, важка за вагою), найбільшого поширення набули пластини, в якості основ яких використовують алюмінієву фольгу або полімери.
Для закріплення сорбенту застосовують гіпс, крохмаль, силиказоль та ін., Які утримують зерна сорбенту на підкладці. Товщина шару може бути різна (100 і більше мкм), але найважливіший критерій - шар повинен бути рівномірний по товщині в будь-якому місці хроматографічної пластинки.
Розчинники
У тонкошарової хроматографії, в якості рухомої фази використовують або чисті речовини (етилацетат, бензол і т.п.), або суміші речовин (системи) у певному співвідношенні.
Підбір рухомої фази (системи) проводиться за такими правилами:
-Вибирають таку систему, в якій компоненти, що розділяються мають невелику розчинність (якщо розчинність речовини висока, то речовини будуть переміщатися з фронтом, при низькій розчинності - залишатися на старті). При розподільної хроматографії або при використанні звернених фаз, розчинність речовин повинна бути вище в рухомій фазі, ніж в нерухомій.
-Склад системи повинен бути постійним і легко відтворюваним.
-Розчинник або компоненти системи не повинні бути отруйними або дефіцитними.
-Система має повністю поділяти речовини близького будови, причому відмінності в Rf повинно бути не менше 0, 05.
-Система не повинна викликати хімічні зміни поділюваних компонентів.
-У вибраній системі аналізовані речовини повинні мати різні значення Rf і розподілятися по всій довжині хроматограми. Бажано, щоб значення Rf лежало в межах 0, 05-0, 85.
-При виборі системи також необхідно враховувати природу поділюваних речовин. Так, при хроматографування речовин, що мають основні властивості система не повинна володіти кислотними властивостями і навпаки.
Тонкошарова хроматографія має кілька способів, пов'язаних, в основному, з видом руху розчинників.
-Висхідна тонкошарова хроматографія
-Низхідна тонкошарова хроматографія
-Горизонтальна тонкошарова хроматографія
-Радіальна тонкошарова хроматографія.
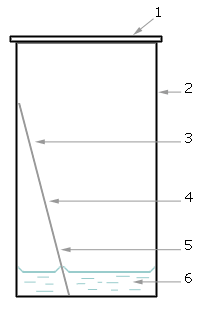
1- кришка
2- скляна камера
3- пластинка ТШХ
4- сорбент
5- місце нанесення проби
6- розчинник
3.3.2 Високоефективна рідинна хроматографія (ВЕРХ).
Високоефективна рідинна хроматографія (ВЕРХ, англійською мовою – HPLC) – це метод розділення речовин, у якому рухомою фазою є рідина, а нерухомою фазою є тонкодисперсна тверда речовина, або рідина, нанесена на твердий носій, або твердий тонкодисперсний носій, хімічно модифікований введенням органічних груп. Розділення речовин у рідинній хроматографії базується на механізмах сорбції, розподілу, іоного обміну або розділення за розмірами молекул. Воно відбувається у колонці рідинного хроматографа, куди під високим тиском подається рідина.
Рідинний хроматограф складається з системи подачі рухомої фази, блока вводу проби, хроматографічної колонки, детектора і реєструючого пристрою. Рухома фаза звичайно подається під тиском із однієї або декількох посудин і протікає через блок вводу проби, колонку, а потім через детектор із заданою швидкістю. Сигнал від детектора перетворюється, підсилюється і реєструється у вигляді хроматограми, аналогічно до хроматограми у газовій хроматографії.
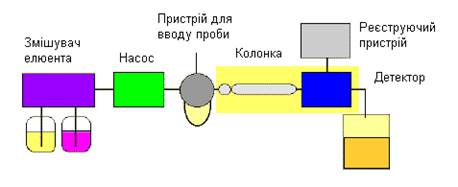
Схема будови рідинного хроматографа
Для вводу проби використовують петлеві дозатори, спеціальні мікрошприци або систему автоматичного пробовідбору.
Рідинний хроматограф – це складніший ніж газовий хроматограф прилад. Це пов’язано з тим, що система подачі елюента містить деякі додаткові вузли: систему дегазації, пристрій для створення градієнту, насоси та вимірювачі тиску. Насоси повинні забезпечити швидкість потоку від 0, 1 до 10 мл/хв при тиску до 400 атм.

Температуру хроматографічної колонки підтримують постійною. Склад рухомої фази може або залишатись постійним протягом всього аналізу (ізократичне елюювання), або може змінюватись відповідно до заданої програми (градієнтне елюювання).
У ВЕРХ звичайно використовують прямі колонки довжиною 10, 15, 25 см з внутрішнім діаметром 4-5, 5 мм. У мікроколонкових хроматографах використовують колонки довжиною 5-6 см і діаметром 1-2 мм. Колонки виготовляють зі скла або нержавіючої сталі.
Колонки заповнюють частинками сорбенту або твердого носія, на поверхню частинок якого нанесена тонка плівка рідкої нерухомої фази. Розмір частинок твердого носія становить 5-10 мкм. Твердий носій готують з поверхнево-пористих матеріалів – силікагелю з привитими на поверхні різними функціональними групами, алюмогелю, пористих стекол, полімерних сорбентів.
Рідка нерухома фаза, нанесена на поверхню твердого носія, становить 0, 75-1, 5 % від маси твердого носія. Звичайно для розділення полярних речовин використовують полярні нерухомі фази і малополярні рухомі фази. Неполярні речовини ділять на неполярних нерухомих фазах з використанням полярних рухомих фаз. Рідкими нерухомими фазами служать речовини різної хімічної природи: гліколі, нітрили, силікони тощо.
У ВЕРХ використовують як нормально-фазовий, так і обернено-фазовий варіанти. У першому випадку полярність нерухомої фази вища за полярність рухомої фази, у другому, навпаки, полярність нерухомої фази є нижчою від полярності рухомої фази.
Для безперервного контролю складу елюату, який витікає з колонки, у рідинній хроматографії звичайно використовують диференційні рефрактометри, фотометричні, УФ-спектрофотометричні, люмінесцентні і кондуктометричні детектори.
Диференційний рефрактометр – це універсальний детектор. Він дозволяє визначити загальний показник заломлення системи проба-елюент, тобто сигнал дають усі компоненти, показник заломлення яких відрізняється від показника заломлення елюенту. Його чутливість» 10-6 г, діапазон лінійності становить 4 порядки. Цей детектор чутливий до зміни температури, тому вимагає термостатування.
УФ-детектор – працює при одній і тій же довжині хвилі, що відповідає найбільш інтенсивній лінії ртутної лампи низького тиску λ = 253, 7 нм. Флуоресцентна приставка дозволяє збуджувати випромінювання з λ = 280 нм. УФ-детектор найбільш чутливим є у випадках, коли молярні коефіцієнти світлопоглинання компонентів високі, а елюент не поглинає в ультрафіолетовій області спектру. У цьому випадку можна використовувати метод градієнтного елюювання. При λ = 254 нм можна визначати будь-які ароматичні сполуки, більшість кетонів та альдегідів (e = 20-104). УФ-детектор селективний, дає можливість визначати 10-9 г, його діапазон лінійності» 5 порядків.
Фотометри та спектрофотометри дають можливість працювати при будь-якій довжині хвилі (190-650 нм), вони реєструють зміну поглинання в часі при певній довжині хвилі або в зупиненому потоці елюенту знімають спектр. Швидкозаписуючий спектрофотометр записує всю спектральну область за 20 с. Спектрофотометричний детектор з лінійкою із 211 діодів на підкладці із кремнію дає можливість вимірювати одночасно велику кількість смуг у вузькому інтервалі довжин хвиль. Отриману інформацію обробляє комп’ютер і зберігає в пам’яті для побудови графіка. Флуоресцентні детектори чутливіші від спектрофотометричних приблизно у сто разів. Їх застосовують для визначення мікродомішок.
Кондуктометричний детектор використовують у іонообмінній хроматографії для вимірювання провідності розчинів (Ом-1), пропорційної до числа іонів у розчині, їх рухливості. Сигнал детектора лінійно залежить від концентрації іонів у широкому інтервалі – від 0, 01 мкг/мл до 100 мкг/мл. Високочутливе кондуктометричне детектування з автоматичним записом сигналу дає межу визначення nּ 10-3 мкг/мл. Використання концентруючої колонки дає можливість знизити межу визначення на 2-3 порядки.
Ідентифікацію речовин у ВЕРХ звичайно проводять одним із таких способів:
- порівняння часу утримування досліджуваної речовини у випробовуваній пробі і розчині порівняння. Розчин порівняння – це розчин чистої досліджуваної речовини, присутність якої передбачається у випробовуваній пробі.
- порівняння відносного часу утримування аналізованої речовини у випробовуваній пробі і розчині порівняння. Відносний час утримування – це відношення часу утримування аналізованої речовини до часу утримування речовини, взятої за стандарт;
- порівняння хроматограми випробовуваної проби з хроматограмою розчину порівняння або хроматограмою, наведеною у окремій статті.
Найчастіше використовують перший спосіб. Другий спосіб доцільно використовувати, якщо умови хроматографування є важко відтворюваними. Третій спосіб доцільно застосовувати для препаратів рослинного і тваринного походження.
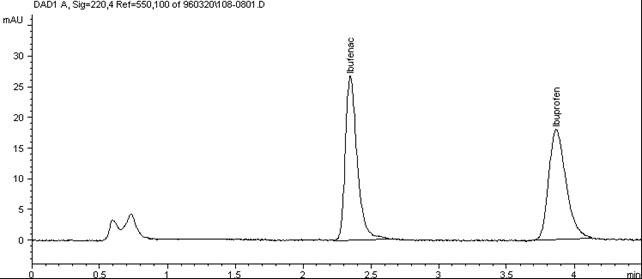 Хроматограма визначення лікарського препарату " Ibuprofen" в сироватці крові людини методом внутрішнього стандарту (внутрішній стандарт – Ibufenac).
Хроматограма визначення лікарського препарату " Ibuprofen" в сироватці крові людини методом внутрішнього стандарту (внутрішній стандарт – Ibufenac).
Кількісне визначення проводять методом абсолютного калібрування та методом внутрішнього стандарту.
Абсолютне калібрування. Цей метод полягає у побудові калібрувального графіку залежності площі піків досліджуваної речовини від її вмісту (концентрації) в пробі. Для цього готують ряд стандартних розчинів з різною концентрацією досліджуваної речовини (не менше 5 розчинів кожної концентрації) і аналізують на хроматографі. Для кожного значення концентрації розраховують середнє значення площі піків і будують градуювальний графік - графік залежності площі хроматографічного піку досліджуваної речовини від концентрації або вмісту цієї сполуки у розчині. Після цього у тих самих умовах хроматографують досліджувану пробу з невідомою концентрацією (декілька паралельних досліджень), вимірюють площу піку досліджуваної речовини і розраховують середні значення площі піків. За градуювальним графіком знаходять концентрацію аналізованої речовини у випробовуваному розчині.
Метод внутрішнього стандарту. Цей метод полягає у внесенні до розчину досліджуваної речовини внутрішнього стандарту. Внутрішній стандарт (речовина-стандарт) – це, як правило, близька за хімічною структурою до досліджуваної речовини сполука, пік якої на хроматограмі знаходиться поруч з піком досліджуваної речовини (рис.4.17)
Градуювальним графіком цього методу є графік залежності відношення площ піків досліджуваної речовини до площ піків внутрішнього стандарту від вмісту (концентрації) досліджуваної речовини в пробі. Для цього готують ряд стандартних розчинів з різною концентрацією досліджуваної речовини (не менше 5 розчинів кожної концентрації) і однакової концентрації внутрішнього стандарту. З цих розчинів готують проби змішуванням однакових об’ємів розчинів досліджуваної речовини і внутрішнього стандарту, які аналізують на хроматографі. Для кожного значення концентрації розраховують середнє значення площі піків і будують градуювальний графік - графік залежності відношення площі піків досліджуваної речовини і внутрішнього стандарту від концентрації або вмісту досліджуваної речовини у розчині. Для випробовуваного розчину пробу готують аналогічно. За градуювальним графіком знаходять концентрацію аналізованої речовини у випробовуваному розчині.
Дані, отримані в результаті аналізу методом ВЕРХ, представляють, вказуючи, звичайно, наступні характеристики: розміри хроматографічної колонки і матеріал, з якого вона виготовлена, тип нерухомої фази, розмір частинок нерухомої фази, температуру колонки, швидкість і склад рухомої фази, тип детектора.
Обов’язковою умовою є попередня перевірка придатності хроматографічної системи для розділення досліджуваної суміші, зокрема:
- відносні часи утримування досліджуваних речовин мають бути близькими до зазначених у методиці величин;
- число теоретичних тарілок (ефективність хроматографічної системи), розраховане за зазначеним піком, має бути не меншим зазначеної величини;
- коефіцієнт розділення зазначених піків, розрахований з хроматограм розчину порівняння, має бути не менше зазначеної величини;
- відносне стандартне відхилення, розраховане для висоти або площі зазначеного піка або їхніх відношень до висоти або площі піка внутрішнього стандарту з хроматограм розчину порівняння, має бути не більше зазначеної величини; для розрахунку відносного стандартного відхилення використовують дані звичайно п’яти паралельних хроматограм.
3.3.3 Адсорбційна хроматографія
В адсорбційному варіанті рідинної хроматографії залежно від полярності нерухомої і рухомої фаз розрізняють нормально-фазну (НФХ) і обернено-фазну (ОФХ) хроматографії. В НФХ використовують полярний сорбент і неполярні рухомі фази, ОФХ – неполярний адсорбент і неполярні рухомі фази. У обидвох випадках вибір рухомої фази є важливішим, ніж вибір нерухомої. Нерухома фаза повинна затримувати досліджувані речовини. Рухома фаза, тобто розчинник, повинна забезпечити ефективне розділення за прийнятний час.
Нерухомі фази. Адсорбенти різних типів (полярні і неполярні) проявляють неоднакову селективність до аналізованих речовин. Як адсорбенти використовують тонкодисперсні пористі матеріали з питомомю поверхнею більше 50 м2/г.
Полярні адсорбенти (силікагель, оксид алюмінію, силікат магнію, оксиди металів) мають на своїй поверхні слабокислі ОН-групи, які здатні утримувати речовини з основними властивостями. Ці адсорбенти використовують головним чином для розділення неполярних сполук і сполук з середньою полярністю.
Недолік полярних адсорбентів – висока чутливість до вмісту води у розчинниках: наприклад, силоксанові групи –Si-O-Si- на поверхні SiO2 у присутності води переходять у силанольні =Si-OН, при цьому міняються властивості поверхні і результати стають невідтворюваними. Для ВЕРХ використовують полярні сорбенти з привитими полярними групами (аміни, діоли та ін.), що дозволяє міняти селективність, підбираючи відповідний елюент.
Неполярні адсорбенти (активоване вугілля, кизельгур, діатоміт) не проявляють селективності до неполярних молекул. Використовують також сорбенти з привитими фазами, наприклад силікагель з алкілсилильними групами від С2 до С22.
Екранувати силанольні ОН-групи силікагелю можна аліфатичними вуглеводнями з С3 і С4, але отримати відносно високі значення ємності і великі значення утримування вдається при введенні довших алкільних ланцюжків, переважно з С18. Такі сорбенти успішно використовуються для визначення багатьох сполук.
Крім названих сорбентів, використовують поверхово-пористі носії (ППН). Це можуть бути жорсткі непористі носії (скляні кульки), покриті тонким пористим шаром активного полярного або неполярного сорбента. Таким сорбентам властивий низький опір потоку, за рахунок чого збільшується швидкість аналізу.
Рухомі фази. У рідинній хроматографії особливо важливий вибір рухомої фази, оскільки вона має значний вплив на селективність розділення, ефективність колонки і швидкість руху хроматографічної смуги. Рухома фаза повинна розчиняти досліджувану пробу, мати малу в’язкість (коефіцієнти дифузії компонентів досліджуваної проби повинні бути достатньо великими), з неї повинно бути можливим виділення розділених компонентів (процес сорбції-десорбції повинен бути зворотнім). Рухома фаза повинна бути інертною у відношенні до матеріалів, з якими вона контактує, безпечною, дешевою, підходити до даного детектора.
Розділення досягають, міняючи елююючу силу рухомої фази – розчинника. Елюююча сила розчинника показує, у скільки разів енергія сорбції даного елюента є більшою, ніж енергія сорбції елюента, вибраного в якості стандарту, наприклад н-гептану. Розчинники (елюенти) ділять на слабкі і сильні. Слабкі розчинники слабко адсорбуються нерухомою фазою, тому коефіцієнти розподілу сорбованих речовин (сорбату) високі. Сильні розчинники сильно адсорбуються, тому коефіцієнти розподілу є низькими. Розчинник тим сильніший, чим вища розчинність у ньому досліджуваної проби, чим сильніша взаємодія розчинник-сорбат.
Є дані про відносну силу розчинників для різних адсорбентів. Для силікагелю сила розчинників збільшується в ряді: пентан (0)< чотирихлористий вуглець (0, 11) < бензол (0, 25) < хлороформ (0, 26) < дихлоретан (0, 32) ацетон (0, 47) < діоксан (0, 49) ацетонітрил (0, 50).
Елюююча сила визначається полярністю розчинника. У нормально-фазовій хроматографії із збільшенням полярності розчинника елюююча сила розчинника росте, в обернено-фазовому варіанті – зменшується. Розташування розчинників відповідно до зростання елююючої сили називається елюотропним рядом. У рідинній адсорбційній хроматографії елюотропний ряд Снайдера має вигляд (у дужках наведено значення елююючої сили): пентан (0)< н-гексан (0, 01)< гептан (0, 01) < циклогексан (0, 04) < чотирихлористий вуглець (0, 18) < бензол < (0, 32) хлороформ (0, 38) < ацетон (0, 51) < етанол (0, 88) < вода.
Для розділення речовин різної полярності і для розділення сполук різних класів речовин використовують нормально-фазову хроматографію: з неполярних рухомих фаз сполуки різних класів виходять із колонки з полярним адсорбентом з різним часом (час утримування сполук з різними функціональними групами збільшується при переході від неполярних сполук до слабополярних). Для дуже полярних молекул tR настільки великий, що при використанні неполярного елюента аналіз неможливий. Для зменшення часу утримування полярних сорбатів переходять до полярних елюентів.
3.3.4 Афінна хроматографія
Назва “афінна хроматографія” була запропонована для відносно нового і ефективного хроматографічного методу, в якому спорідненість відіграє особливо важливу роль. Описуваний метод базується на специфічній взаємодії, характерній для деяких біологічних та біохімічних процесів. Ця взаємодія відбувається між парами речовин, які реагують в розчині з високою вибірковістю. Так, наприклад, антиген та антитіло специфічно зв’язуються між собою, фермент реагує тільки зі своїм субстратом або з інгібітором, транспортна рибонуклеїнова кислота “вибирає” тільки ту амінокислоту. яку вона можк перенести всередину рибосоми, гормон реагує з відповідним рецептором. Якщо одна із цих сполук. яка відноситься до будь-якої з перерахованих пар, утримується ковалентним зв’язком на відповідному носії і при цьому зберігає свої властивості (специфічні характеристики), то подібний десолюбілізований препарат можна використовувати для вибіркового зв’язування із розчину другої сполуки пари. Якщо перемішати десолюбілізований препарат (у вигляді частинок з розчином суміші декількох речовин, то при цьому препарат зв’язує тільки один певний компонент. Отриманий препарат звільняють від супутніх речовин фільтруванням або центрифугуванням. промивають і відповідним чином виділяють необхідну сполуку.
Якщо ввести десолюбілізований препарат в хроматографічну колонку і повільно пропускати через неї розчин, то потрібний компонент зв’яжеться і буде утримуватись у колонці. Після промивання колонки відповідною десорбуючою рідиною можна вибірково елюювати цей компонент. За своїм апаратурним оформленням і за методикою