Главная страница Случайная страница
КАТЕГОРИИ:
АвтомобилиАстрономияБиологияГеографияДом и садДругие языкиДругоеИнформатикаИсторияКультураЛитератураЛогикаМатематикаМедицинаМеталлургияМеханикаОбразованиеОхрана трудаПедагогикаПолитикаПравоПсихологияРелигияРиторикаСоциологияСпортСтроительствоТехнологияТуризмФизикаФилософияФинансыХимияЧерчениеЭкологияЭкономикаЭлектроника
Механизм сопряжения дыхания и фосфорилирования в митохондриях 5 страница
|
|
Фосфоглицериды клеточных мембран гидралнзуются с помощью фосфо- липаз А), А2, С и D, локализованных преимущественно в лизосомах. Но отдельные фосфолипазы имеются и в других органоидах клетки. Продуктами гидролиза фосфоглицеридов являются глицерин, 'жирные кислоты, азотистые спирты, неорганический фосфат. Имеются специфические ферменты гидролиза сфинголипидов и гликолипидов, которые участвуют в их обновлении.
Известно, что гидролиз внутриклеточных липидов ие приводит к накапли- нию глицерина и жирных кислот. Это говорит о том, что скорость гидролиза сбалансирована со скоростью их окисления внутри клетки. В жировой ткани образующиеся в результате гидролиза триацилглицеринов глицерин и жирные кислоты не подвергаются окислению, а поступают в кровь, из которой потребляются другими органами.
Окисление глицерина
Обмен глицерина тесно связан с гликолизом, в который вовлекаются метаболиты глицерина по следующей схеме:
HjC—он н, с—он н2с—он
НС—ОН ГЛ|" И^*> С*^*Ц*. —он «-гя»ц«ро< фос».тип1дрогся.м ^ с==ю
hjc—он Hji—opo3h1 ная+ н»я-н+н* hjc_opo3h2
пмиирин а ттшю**к«шт ыпиоктиипв-
| 9* |
Сначала глицерин при участии глицеролфосфокиназы. превращается в о-гли- церолфосфат. Последний под действием НАД-зависимой а-глииеоолфосфат- дегидрогеназы превращается в дигидроксиацетонфосфат, который, являясь обычным метаболитом гликолиза, включается в гликолиз и превращается его ферментами до лактата в анаэробных условиях или до С03 и HzO в аэробных. Превращение одной молекулы глицерина дает одну молекулу АТФ в анаэробных условиях и 19 молекул АТФ в аэробных. Глицерин — хороший энергетический субстрат и используется в этих целях практически всеми органами и тканями.
Окисление жирных кислот
(Окисление высших жирных кислот было впервые изучено в 1904 г. Кноопом, который, вводя животным фенилзамещенные жирные кислоты, показал, что в ходе их окисления происходит постепенный отрыв двух углеродных фрагментов с, карбоксильного конца. Кнооп назвал механизм окисления жирных кислот Р-окислением. В 1948—1949 гг. Кеннеди и Ленинджер установили, что окисление жирных кислот происходит только в митохондриях. Лннен с сотр. (1954—1958) описал основные ферментативные процессы окисления жирных кислот. В настоящее время р-окисленне жирных кислот называют циклом Кноопа—Лннйна.
Жирные кйслоты, образовавшиеся в клетке путем гидролиза триаинлглн- церинов или поступившие в нее из кровн, должны быть активированы. Активирование их происходит в цитоплазме с участием ацил-КоА-синтетазы по схеме
C^-fCHjK-CHj-CHsr-COOH + АТФ + KoASH
О
II
-► CHj— (СН,), —CHs—CHj—C~SKoA + АМФ + H, P20,
Поскольку этот процесс идет вне митохондрий, то далее необходим транспорт ацила через мембрану внутрь митохондрий.
Транспорт происходит с участием карнитина, на который перебрасывается ацнл с ацил-КоА на внешней стороне. Ацилкариитии диффундирует к внутренней стороне мембраны, где отдает свой ацнл КоА, находящемуся в матриксе. Процесс обратимого переноса ацила между КоА и карнитином на внешней и внутренней стороне мембраны осуществляется ферментом ацил-КоА-карни- тингтрансферазой (рис. 52).
Рис. 52. Схема транспорта жирных к
В матриксе происходит окисление жирной кислоты в цикле Кноопа— Лннена. В состав этого цикла входят четыре фермента, которые последовательно действуют на ацил-КоА. К" ним относятся: ацил-КоА-дегидрогеназа
| „-< „4- |
(ФАД-зависимый фермент),
еноил-КоА-гидратаза, 3-гидро- ксиацил-КоА-дегидрогеназа (НАД-зависимый фермент) и ацетил-КоА-ацилтрансфераза. За один виток или оборот цикла от жирной кислоты отрывается остаток уксусной кислоты в виде ацетил-КоА и образуется одна молекула ФАД-Н2 и одна молекула НАД • Н2 (рис. 53). Затем циклы повторяются до тех пор, пока жнрная кислота не укоротится до четырехугле- родного фрагмента —. бутирнл- КоА. На последнем витке бутирил-КоА разрывается пополам, поэтому образуется не одна, а две молекулы ацетнл- КоА.
| г через митохондр»альную мембрану. |
| Цитоплазма Мембрана |
| Митохондрий Матрикс НО—Карнитин |
Продуктами окисления жирной кислоты с четным числом углеродных атомов являются ацетил-КоА, ФАД• Н2 и НАД- Н2. Далее ацетил-КоА вступает в цикл Кребса, а ФАД-Н2 и НАД-Н, — прямо в дыхательную цепь.
Особенности окисления жирных кислот с нечетным
'" " Г
числом углеродных атомов состоят в том, что наряду с обычными продуктами окисления (как у четных) —ацетил-КоА, ФАД.Н2и НАД-Н2, образуется одна молекула пропионил-КрА (СН3—СН2—CO~SKoA) на молекулу —окисленной жирной кислоты. Пропионил-КоА превращается в сукцинил-КоА: со, j: ooH
СН, —сн, —co~sxoa ---- —си, —сн—co-skoa»- ноос—сн, —сн, —с o-skoa
Карбоксилирование пропионил-КоА осуществляется под действием пропионил- КоА-карбоксилазы. (коферментом этого фермента служит биотин—переносчик ка^биКеш рупп, уеакиин ipeoyei 1акж«г АТФ). Образовавшийся метилмало- нил-КоА превращается метилмалонил-КоА-мутазой (кофермент которой де- зоксиаденозилкобаламин является производным внтамнйа В12) в сукцинил- КоА, который вступает.в цикл Кребса.
Особенности окисления ненасыщенных жирных кислот определяются положением и числом двойных связей в их молекулах. До места двойной связи ненасыщенные жирные кислоты окисляются так же, как насыщенные. Если двойная связь имеет ту же конфигурацию (транс-конфигурацию) и расположение (Д[5]-3), что и в еноил-КоА, образующемся при окислении насыщенных жирных кислот, то далее окисление идет обычным путем. В противном случае в реакциях участвует дополнительный фермент А3-*-цие-А2-3-транс- еноил-КоА-изомераза, который способствует перемещению двойной связи в нужное положение и изменяет конфигурацию ее из цис- в транс-.
Скорость окисления ненасыщенных жирных кислот выше, чем насыщенных. Например, если взять за эталон скорость окисления насыщенной стеариновой кислоты, то скорость окисления олеиновой в 11, лизолевой в 114, лино- леяовой в 170, а арахидоновой почти в 200 раз выше, чем стеариновой'.
Кроме р-окисления встречаются еще два пути окисления жирных кислот, получившие название а- и ©-окисления. Они малоактивны и связаны с образованием сначала а- и ш-гидроксикислот, а затем с их дальнейшими превращениями. Эти пути окисления не имеют такого энергетического значения, как.^•окисление, и, возможно, связаны с какими-то специальными функциями клетки.
Энергетический баланс окисления жирных кислот. Энергетическая- ценность жирной кислоты с четным числом углеродных атомов рассчитывается следующим образом. Если жирная кислота содержит 2п атомов углерода, то при полном ее окислении образуется п молекул ацетил-КоА (каждый ацетил имеет два атома углерода) и п—1 молекул ФАД-Н2 и НАД-Н2 (поскольку в ходе последнего цикла окисления образуется 2 молекулы ацетил-КоА, но по одной молекуле ФАД • Н2 и НАД • Н2). Окисление ФАД ♦ Н2 дает 2 АТФ, а НАД» Hj — 3 АТФ; вместе они дают 5 АТФ или в общем виде 5(л— 1) АТФ. Полное сгорание одной молекулы ацетил-КоА, как уже говорилось, приводит к образованию 12 молекул АТФ, а п молекул ацетил-КоА дают 12л молекул АТФ. Одна молекула АТФ затрачивается на активирование жирной кислоты, следовательно, остается 12л—1 молекул АТФ. В итоге баланс АТФ при полном окислении жирной кислоты с четным числом атомов углерода можно выразить формулой
1) + 12л— 1 = 17л—6 «оявкул АТФ.
где п равно половине числа атомов углерода, содержащихся в конкретной жирной кислоте. Например, молекула пальмитиновой кислоты, содержащая 16 атомов углерода, дает 130 молекул АТФ.
Энергетическая ценность жирных кислот выше, чем, например, глюкозы. Так, полное сгорание капроновой кислоты, имеющей то же число атомов углерода, что и глюкоза, дает 45 молекул АТФ (глюкоза дает 38 молекул АТФ). Однако для сгорания в цикле Кребса молекул ацетил-КоА, образующихся при 0-окислении, требуется достаточное количество оксалоацетата-. В этом отношении углеводы имеют преимущество перед жирными кислотами, ибо при их распаде образуется пируват, являющийся источником образования не только ацетил-КоА, но и оксалоацетата (нируваткарбоксилазная реакция;, т. е. облегчается превращение ацетил-КоА в цикле Кребса. Не случайно в биохимической литературе бытовало выражение, что «жиры сгорают в пламени углеводов», поскольку образующийся уже в гликолизе АТФ может использоваться для активирования жирных кислот в цитоплазме, а образующийся из пирувата оксалоацетат облегчает включение ацетильных остатков жирной кислоты в цикл Кребса.
Значение жирных кислот как энергетических субстратов для разных органов и тканей. Не все ткани с одинаковой интенсивностью используют жирные кислоты и промежуточные продукты их окисления —- кетоновые тела — как энергетические субстраты. Активно используют жирные кислоты сердце, а также почки, скелетные мышцы (при длительной работе). В этих же органах сгорают кетоновые тела, которые служат источником энергии. В нервной ткани доля использования жирных кислот и кетоновых тел для обеспечения энергетических нужд незначительна.
собой циклический процесс, который протекает на поверхности пальмитатсннтетазы.
Образование малонил-КрА для синтеза жирных кислот. Субстратом для образования малонил-КоА служит ацетил-КоА. Имеется несколько путей поступления ацетил-КоА в цитоплазму. Один из них — перенос ацетильных остатков нз матрнкса митохондрий через митохондриальную мембрану в цитоплазму. Этот процесс сходен с переносом жирных кислот и осуществляется также с участием карнитина и фермента ацетил-КоА-карнитинтрансферазы. Другой путь — это образование ацетил-КоА из цитрата. Цитрат поступает из митохондрий и в цитоплазме расщепляется с помощью фермента АТФ- цитрат лиазы:
Цитрат + А 1< Р + КоА ► Ацетил-КоА + Оксалоацетат + АДФ + Ф„
Реакция практически необратима и сдвинута вправо.
Из ацетил-КоА, поступающего этими путями в цитоплазму, образуется малонил-КоА:
сн3 соон
с=о +нсоэ + атф ■ „/У" -> сн, +адф+н.ро4
■ I (Е'биотип)!
SKoA с_0
ацетнл-КоА I
SKoA
Реакция катализируется биотиновым ферментом ацетил-КоА-карбоксилазой (Е-биотин) с участием ионов Mg2+. Этот фермент является тетрамером с молекулярной массой 400 000—500 000.
Стадии синтеза жирной кислоты на поверхности пальмитатсннтетазы. Пальмитат-синтетаза состоит нз семи ферментов, каждый из которых выполняет определенные функции. В центре полиферментного комплекса находится ацил-переносящий белок (АПБ), а по периметру шесть остальных ферментов. АПБ выполняет роль акцептора и распределителя ацильных остатков. В егс составе имеется ковалентно связанный 4-фосфопантетеин, содержащий свободную SH-группу, с которой н связываются ацилы. Кроме этой центральной SH-группы у пальмитатсннтетазы имеется и периферическая. Обе SH-группы как акцепторы ацнлов участвуют в процессе синтеза жирных кислот на поверхности полиферментного комплекса. SH
Если пальмитатсинтетазу обозначить символом, то циклический
процесс синтеза жирной кислоты можно описать рядом последовательно протекающих реакций.
1. Перенос ацетила с ацетил-КоА на синтетазу: о
СН3 II
SH 1, s~c—сн3
Е< Е< + KoASH
SH (NSH
SKoA
Эта реакция осуществляется первым ферментом пальмитатсннтетазы — ацетилтрансацилазой, имеющей SH-группу. Ацетил здесь выполняет роль затравки в синтезе.
2. Перенос малонила с малоиил-КрА на синтетазу:
О СООН о
II I II
S~C—CHs < *на S~C—СН,
/ I /
Е v г С==0 — Е О + KoASH
NSH I \ ||
& КоА S~C—сн2—соон Реакция осуществляется вторым ферментом синтетазы—малонилтрансаци- лазой.
3.Конденсация ацетила с малонилом и декарбоксилирование образовавшегося продукта:
О
II
S-C—СНЭ SH
Е ^ О О О + с°2
\ II \ -И II
S~C—СНг- СООН S~C—CHj-C—сна
Реакция катализируется третьим ферментом синтетазы — $-кетоацил-синтета- зой. При этом образуется ацетоацетил, связанный с сиитетазой.
4. Первое восстановление промежуточного продукта с участием НАДФ • Н2 , SH SH
Е/ О О в' О ОН
^е — Г—ГН.—Г—ГН..J. _
НАДФН+Н + НАДФ+
Реакция катализируется четвертым ферментом синтетазы — $-кетоацил-ре- дуктазой с образованием гидроксибутирила. 5. Дегидратация промежуточного продукта:
Е О ОН Е О +Н20
\ 1! | \ и
S—С—СН2—СН—СН, S~C—СН=СН—СН3
Реакция катализируется пятым ферментом синтетазы — гидроксиацил-гидра- тазой с образованием кротоннла.
6. Второе восстановление промежуточного продукта с участием НАДФ • Нг
Ч - с сн=сн сн3 ндд^н, " > лд4, Ч~1-сн1-сн, -сн,
Реакция катализируется шестым ферментом синтетазы — еноилредуктозой с образованием бутирила, связанного с ферментом. Синтезированный бутирил переносится с участием первого фермента синтетазы — ацетилтрансацилазы — на ту SH-группу (на схеме верхняя), с которой вначале был связан затравочный ацетил. На освободившуюся SH-группу (нижнюю) поступает новый малонильный остаток:
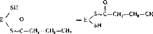
|
Цикл синтеза повторяется.
Для синтеза пальмитиновой кислоты нужно семь таких циклов, соответственно требуется семь остатков малонила и один ацетил. Последний является концевым фрагментом жирной кислоты. Синтезированная пальмитиновая кислота или переносится на внешний КоА с образованием ацил-КоА, или, чаше, гидролизуется с помощью специфической пальмитатдеацилазы с образованием свободной жирной кислоты.
Удлинение жирных кислот. В митохондриях и эндоплазм этической сети возможно удлинение синтезированных или поступивших с пищей жирных кислот. Этот процесс отличается от синтеза жирных кислот заново. В мито, - хондриях удлинение осуществляется с помощью комплекса ферментов путем добавления ацетильных остатков от ацетил-КоА. В эндоплазматической сети удлинение осуществляется комплексом ферментов путем использования малой ил-Ко А.
Н—с—ОН
 глицерол фосфат
глицерол фосфат
|
Биосинтез ненасыщенных жирных кислот. В тканях млекопитающих возможно образование только моноеновых жирных кислот. Из стеариновой образуется олеиновая, из пальмитиновой — пальмитоолеиновая. Этот синтез происходит в эндоплазматической сети клеток печени с помощью монооксигеназ- ной цепи окисления. Остальные ненасыщенные жирные кислоты не образуются в организме человека и должны поступать " с растительной пищей {в растениях образуются полиненасыщенные жирные кислоты). Полиненасыщенные жирные кислоты для млекопитающих являются незаменимыми факторами пищи.
Биосинтез три ацнл глицеринов
Синтез триацилглнцеринов происходит при депонировании липидов в жировой ткани или в других тканях организма. Этот процесс локализуется в гиалоплаз- ме клеток.
Непосредственно для синтеза триацилглнцеринов используется а-гяице- ролфосфат, а не свободный глицерин, и ацил-КоА, а не свободная жирная кислота, а-Глицеролфосфат образуется путем фосфорилирования поступающего в ткани глицерина или при восстановлении промежуточного продукта гликолиза дигидроксиацетонфосфата.
Первой стадией синтеза 'триацилглнцеринов служит образование фосфа- тндной кислоты с участием глицерофосфат-ацилтрансфераэы:
 Н2С—ОРО, Н,
фэсфаткан» кислота
Н2С—ОРО, Н,
фэсфаткан» кислота
|
Далее фосфатидная кислота подвергается действию фосфатидат-фосфатазы с образованием диацилглицерина:
Н2С—о—С—R нгс—О—с—R
НС—О—С—R' —НС—О—С—R' i -н, ро. |
HjCOPOjHj
фосфатная
На диацилглицерин с помощью диацилглицерол-ацилтрансферазы. переносится третий ацильный остаток:
Н2С—О—С—R
r" —со—sk о а
Синтезируемый триацилглицерин накапливается в виде жировых включений в цитоплазме клеток.
Биосинтез фосфолипидов
Синтез фосфолипидов связан с обновлением мембран. Этот процесс протекает в гиалоплазме тканей. Первые стадии.синтеза фосфолипидов и триаынлглице- ринов совпадают. Эти пути расходятся на уровне фосфатидной кислоты и диацилглицерина {рис. 54).
Существует два пути синтеза фосфолипидов, причем для обоих необходим ЦТФ. Первый путь связан с вовлечением фосфатидной кислоты в синтез фосфоглицеридов. Взаимодействие ее с ЦТФ приводит к образованию ЦДФ- диацилглицернна, который как кофермент способен участвовать в переносе диацилглицерина на серин {или инозит). При этом образуется фосфатидил- серин {или фосфатидилинозит). Серинфосфатиды декарбоксилируются (коферментом служит пиридоксальфосфат) и образуются этаноламинфосфатиды. Последние метилируются с участием S-аденозилметионина {донор трех метальных групп), а переносчиками метильных групп служат тетрагидрофолневая кислота и метилкобаламин.
Второй путь синтеза связан с активированием спирта, например холина, с образованием ЦДФ-холина. Последний участвует в переносе холина на диацилглицерин с образованием фосфатиднлхолина.
Синтезированные фосфолипиды переносятся с помощью липидперенося- щих белков цитоплазмы к мембранам (клеточным, внутриклеточным) и встраиваются на место старых молекул.
Биосинтез фосфоглвдерплоп Вносите} тряапнлглносранов Биосинтез фосфоглнеродоа (2-й путь) (1-й путь)
Фосфатидил инозит
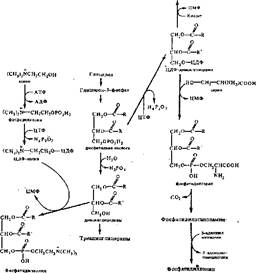
|
Рис. 54. Схема двух путей синтеза некоторых фосфолнпидов (по Т. Т. Березову и Б. Ф. Коровкииу)
Вследствие конкуренции между путями синтеза фосфолнпидов и триацилглицеринов за общие субстраты все вещества, способствующие синтезу фосфолнпидов, препятствуют отложению триацилглицеринов в тканях. Эти вещества называются липотропными факторами. К ним можно отнести структурные компоненты фосфолнпидов — холин, инозит, серии; вещество, облегчающее декарбоксилирование серинфосфатидов — пиридоксальфосфат; донор метиль- ных групп — метионин; фолиевую кислоту и цианкобаламин, участвующих в образовании коферментов переноса метальных групп (ТГФК и метилкобала- мин). Их можно использовать как лекарственные препараты, препятствующие избыточному отложению триацилглицерина в тканях (жировая инфильтрация).
Биосинтез кетоновых тел
Кетоновыми, или ацетоновыми, телами называются три вещества: ацетоаце- тат, p-гидроксибутират и ацетон. Они являются недоокисленнымн, промежуточными продуктами распада, главным образом жирных кислот и углеродных склетов так называемых кетогенных аминокислот (лейцин, изолейции, лизин, фенилаланин, тирозин и триптофан). Образование кетоновых тел, или кето- генез, происходит в митохондриях печени (в других органах кетогенез отсутствует). Возможны два пути кетогенеза. Наиболее активный из них — это гидроксиметилглутаратный цикл, названный так по ключевому соединению, образующемуся в данном цикле. Другой — деацилаэный. Он малоактивен. Исходным веществом для биосинтеза кетоновых тел служит ацетил-КоА.
Гидроксиметилглутаратный цикл. На первой стадии происходит конденсация двух молекул ацетил-КоА с участием ацетил-КоА-ацетилтрансферазы:
СН3—C~SKoA + CH3—e~SKoA — СН, —С—СН, —C~SKoA+ KoASH
II II II II
О О 0 0
Далее ацетоацетил-КоА конденсируется еще с одной молекулой ацетил-КоА с участием гидроксиметилглутарил-КоА-синтазы:
СН8
[
СНа—С— С Н2—С~ S КоА + СН, —C~SKoA — НООС—СНг— С—СН, —C~SKoA + KoASH
p-Гидрокси-р-метилглутарил-КоА расщепляется под действием гидроксиметил- г$утарил-КоА-лиазы на ацетил-КоА и ацетоацетат:
СН3
НООС— CHj—С—CHj— C~SKoA -»■ CHj—C~SKoA + НООС—СН, —С—CH3
Ацетил-КоА вновь используется на первой стадии и тем самым замыкает процесс в цикл. Ацетоацетат — представитель семейства кетоновых тел, $ конечным продуктам гидроксиметилглутаратного цикла.
Остальные кетоновые тела образуются из ацетоацетата: р-гндроксибути- рат — путем восстановления его с участием НАД-зависимой гидроксибутират- дегидрогеназы, а ацетон — в результате декарбоксилирования ацетоацетата
| S |
| НООС—ГН —Г—сн3 |

|
|
| над-*" |
| над h+h" |
ноос—сн2—СН—СН-
Н3С—С—СН, б
с участием ацетоацетат-декарбоксилазы.
Деацилаэный путь кетогенеза возможен после образования ацетоацетил- КоА, который в печени с участием ацетоацетил-КоА-гидролазы, или деацила- зы, гидролизуется'до ацетоацетата.
В печени кетоновые тела далее не превращаются, а поступают в кровь. В норме содержание кетоновых тел (в виде ацетоацетата и р-гидроксибути- рата) составляет всего 0, 1—0, 6 ммоль/л. Остальные ткани и органы (сердце, легкие, почки, мышцы и даже нервная ткань) в отличие от печени используют их как энергетические субстраты. В клетках этих тканей ацетоацетат и р-гид- роксибутират в конечном счете включаются в цикл Кребса и «сгорают» до СОг и Н20 с выделением энергии.
Биосинтез холестерина
В опытах с радиоактивно меченной уксусной кислотой, которая скармливалась животным, было установлено, что углеродный скелет холестерина целиком состоит из углерода уксусной кислоты.
Биосинтез холестерина из ацетнл-КоА происходит с участием ферментов эндоплазматической сети и гиалоплазмы многих тканей и органов. Наиболее активен этот процесс в печени взрослого человека.
Биосинтез холестерина — многостадийны-й процесс, который можно разбить на три этапа:
1) образование мевалоновой кислоты из ацетил-КоА;
2) синтез из мевалоновой кислоты «активного изойрена» с конденсацией последнего в сквален;
3) превращение сквалена в холестерин.
Начальные реакции первого этапа до образования р-гидрокси-0-метил- глутарнл-КоА из ацетил-КоА сходны с начальными реакциями кетогенеза с той лишь разницей, что кетогенез протекает в митохондриях, а биосинтез холестерина — вне митохондрий:
2 Ацетил - КоА--------- А аетоацеткл - КоА! Ацетил - КоА»- 0 • Ги дроке н - g - м ети л гл ут а ри л - Ко А
Далее р-гидрокси-р.метилглутарИл-КоА под действием гидроксиметилглуто- рил-КоА-редуктазы превращается в мевалоновую кислоту:

|
| 2НЛДФ |
Г'
НООС—CHj— С—CHj—С-SKOA ОН о
сн, 1 '
НООС—СН, —С—CHj—CHjOH + KoASH
I
ОН
Эта реакция необратима и лимитирует скорость биосинтеза холестерина в целом.
Возможен и второй путь образования мевалоновой кислоты, отличающийся от описанного тем, что образование р-гидроксн-р-метилглутар ильного остатка происходит на поверхности ацилпереносящего белка {как при биосинтезе жирных кислот). Промежуточный продукт этого пути р-гидрокси-р-метил глутарил-Б-АПБ восстанавливается другим ферментом до мевалоновой кислоты.
На втором этапе мевалоновая кислота в ходе нескольких ферментных реакций, в которых расходуется АТФ, превращается в изопентилпирофосфат и его изомер 3, 3-диметилаллилпирофосфат. Оба эти вещества н есть «активный изопрен», который используется на образование сквалена.
На третьем этапе из сквалена образуется холестерин:
Сквален—* Ланостерин—»■ Холестерин
Гидроксилирование' стероидного кольца протекает с участием монооксигеназ- ной цепи мембран эндоплазматической сети.
Эфиры холестерина образуются путем переноса ацила с ацил-КоА или с фосфатидилхолина на гидроксильную группу холестерина. Последний процесс катализируется фосфатидилхолин-холестерин-ацилтрансферазой:
Фосфатидилхолиы + Холестерин ► Лиэофосфатндилхолнн + Эфир холестерина
Эфиры холестерина особенно активно образуются в слизистой кишечника и печени.
Таким образом, холестерин в тканях может синтезироваться из любых веществ, при распаде которых образуется ацетил-КоА. К ним относятся углеводы, аминокислоты, жирные кислоты, глицерин.
Печень играет главную роль в обмене холестерина. В ней синтезируется до 90% всего эндогенного холестерина и его эфиров; она же участвует в распределении его по другим органам (так как в печени происходит синтез апо- протеинов пре-р-липопротеидов, а-липопротеидов и p-липопротеидов, транспортирующих с кровью холестерин) и в выделении холестерина с желчью. Кишечной микрофлорой некоторая часть холестерина разрушается, большая часть восстанавливается до копростанола и холестанола, которые вместе с небольшим количеством неизмененного холестерица выделяются с калом.
Холестерин, преимущественно в этерифицированном виде, используется на построение биомембран в клетках. Кроме того, из холестерина образуются важные в биологическом отношении стероидные соединения: желчные кислоты (в печени), стероидные гормоны (в коре надпочечников, женских и мужских половых железах, плаценте) и витамин D3, или холекальциферол (в коже).
3. Регуляция обмена липидов в организме
Интенсивность превращения липидов в тканях организма зависит от поступления липидов с пищей и нервно-гормональной регуляции. Избыточное по
требление калорийной пищн — углеводов, триацилглнцеринов, препятствует расходу эндогенных запасов триацилглнцеринов в жировой ткани. Более того, углеводы служат прекрасным источником новообразования различных липидов, поэтому прием большого количества только углеводистой пищи оказывает существенное влияние на образование триацилглицеринов и холестерина.
Синтез эндогенного холестерина также регулируется поступающим с пищей экзогенным холестерином: чем больше потребляется с пищей холестерина, тем меньше его образуется в печени. Экзогенный холестерин тормозит активность гидроксиметилглутарил-КоА-редуктазы и циклизацию сквалена в ланостерин.
Существенную роль в превращении липндов в организме играет соотношение в пище различных липидов. От количества полннгпасыщенлых жирных кислот н фосфолипидов зависит не только всасывание жирорастворимых витаминов, растворителями которых они являются, но и растворимость и стабильность холестерина в жидкостях организма (плазме крови, лимфе) и желчевы- водящих путях. Растительные масла, содержащие много фосфолипидов и полненовых кислот, препятствуют избыточному накоплению холестерина, его отложению в сосудах н других тканях и способствуют выведению его из организма. Наиболее сильное влияние на эти процессы оказывают кукурузное, сафлоровое, хлопковое и подсолнечное масла. Потребление ненасыщенных жирных кислот, имеющихся в растительных маслах, оказывает благоприятное воздействие на синтез эндогенных фосфолипидов, субстратами которых' они являются, и на образование других веществ, для которых требуются полие- новые жирные кислоты, например простагландинов. Являясь разобщитеяямн окислительного фосфорилирования, ненасыщенные жирные кислоты ускоряют процессы окисления в митохондриях тканей и тем самым регулируют избыточное отложение триацилглнцеринов.
Существенное влияние на биосинтез фосфолипидов и триацилглнцеринов оказывают липотропные факторы. Как уже говорилось, они облегчают биосинтез фосфолипидов. Отсутствие их в пище способствует образованию триацилглнцеринов.
Голодание вызывает мобилизацию трнацнлглицеринов из жировой ткани и угнетает эндогенный биосинтез холестерина из-за малой активности гид- роксиметилглутарил-КоА-редуктазы. Последнее открывает путь для активного производства кетоновых тел в печени.
Нервно-гормональная регуляция' липидного обмена сказывается в основном на мобилизации и синтезе триацилглнцеринов в жировой ткани. Липолиз в тканях зависит от активности триацилглицеринлипаэы. Все регуляторы, способствующие переходу неактивной (иефосфорилированной) липазы в активную (фосфорилированную), стимулируют липолиз и выход жирных кислот в кровь. Стимуляторами этого процесса являются адреналин и норадреналин (выделяющиеся в окончаниях Симпатических нервов), гормоны (глюкагон, адреналин, тироксин, трннодтнронин, соматотропин, p-липотропин, кортико- тропин и др.), межтканевые регуляторы, или гормокоподобкые вещества (гистамин, серотонйн и т. д.). Инсулин, наоборот, угнетает аденилатцнклазу, чем препятствует образованию активной лнпааы в жировой ткани, т. е. тормозит липолиэ. Кроме того, инсулин способствует новообразованию триацилглнцеринов из углеводов, что в целом обеспечивает отложение липидов в жировой ткани, а также образование холестерина в других тканях. Гормоны щитовидной железы тироксин и трииодтиронин способствуют окислению боковой цепи холестерина и выведению холестерина с желчью в кишечник.